摘要
禽致病性大肠杆菌(APEC)是导致雏鸡大肠杆菌病爆发的原因,导致急性至过急性死亡。大肠杆菌病引起的高发病率和死亡率导致世界范围内动物福利差、可持续性降低和经济损失。为了进一步了解APEC的分子流行病学、基因组相关性和毒力特征,我们在2018-2021年期间对45个确诊的第一周死亡率(FWM)高的大肠杆菌肉鸡群进行了系统采样。对219株APEC分离株进行了全基因组测序和生物信息学分析。生物信息学分析包括序列分型(ST)、血清分型、毒力相关基因(VAGs)检测和系统发育分析。结果表明,ST23、ST429和ST95在来自挪威肉鸡群的APEC分离株中具有较高的流行率,与ST95、ST69和ST10相比,ST23、ST429、ST117和ST371更容易单独致病。系统发育分析,连同相关的元数据,确定了由ST429和ST23引起的两次不同的大肠杆菌病爆发,并深入了解了具有相同st的鸡群内部和之间的预期SNP距离。此外,我们的结果强调需要结合两种分型方法,如血清分型和序列分型,以更好地区分APEC菌株。最终,从一群鸟中对多只鸟进行APEC的系统采样,并将WGS作为诊断工具,对于确定一群鸟中引起疾病的APEC和发现整个农场的大肠杆菌病爆发非常重要。
介绍
大肠杆菌病被认为是禽类生产中一种非常重要的疾病,因为它在世界范围内造成动物福利不良和巨大的经济损失。它是由禽致病性引起的大肠杆菌(APEC),它被归类为肠道外致病菌大肠杆菌(ExPEC),以及新生儿脑膜炎大肠杆菌(NMEC)和尿路致病性大肠杆菌(UPEC)。当鸡群中雏鸡的死亡率在孵化后第一周迅速上升,通常在孵化后2至5天达到高峰时,可怀疑大肠杆菌病。在尸检中,雏鸡表现出典型的与急性至急性多浆液炎相关的宏观病变,如脾脏肿大,浆液膜和脐部水肿。在疾病的后期,大肠杆菌血症可导致纤维性多浆膜炎,纤维蛋白渗出物覆盖内脏器官的浆膜,如肝脏(肝炎周围)和心脏(心包炎)。分离鉴定大肠杆菌确认诊断[1]。
消化系统致病性大肠杆菌,包括APEC,被认为是一组不同的病原体,一些研究试图根据毒力相关特性对APEC进行分类和鉴定[2,3.,4]。从历史上看,大肠杆菌已通过检测体细胞o抗原与抗血清进行血清分组。最常见的APEC血清群是O1、O2和O78 [1,5]。后来,系统分组,用三重PCR方法进行分组大肠杆菌根据三个基因的存在与否来划分大肠杆菌分为7个类群:A, B1, B2, C, D, E和F,已使用[6]。MLST是另一种常用的类型方法大肠杆菌,包括亚太经合组织(APEC)在内,它是基于人类基因组中七个管家基因的组合大肠杆菌基因组(7]。在欧洲最常见的APEC标准包括ST23、ST69、ST95、ST117、ST131、ST140及ST428/429 [5]。
另一方面,全基因组测序(WGS)实现了一系列计算机鉴定方法,如MLST、血清分型、VAGs鉴定和系统发育分析[5,8]。核心基因分析和系统发育分析,可在核苷酸水平上检测变异,并可用于研究相关性,后者可用于检测和确认可能的疫情及其起源。然而,系统发育方法在不断发展,这种分子分析的结果需要深入了解基因组,基本的生物信息学知识以及对待研究病原体的了解[9,10]。
2014年至2016年期间,包括挪威在内的北欧国家被诊断为大肠杆菌病的鸡群突然增加[11]。对感染禽的分离株进行全基因组测序和基因组调查,发现ST117 O78:H4的主要谱系,但也存在遗传多样性群体。这一经验强调了在挪威肉鸡生产中需要更多的APEC分子流行病学知识。因此,2018年开始对第一周死亡率高的肉鸡群进行系统抽样。
本研究的目的是系统地收集高FWM挪威肉鸡群的APEC分离株,并利用WGS和生物信息学分析对同一鸡群和鸡群之间的分离株进行深入的表征和比较。每次采样都收集了与种群相关的元数据,如采样日期、孵化场、亲本种群和杂种,从而能够检测所识别的APEC类型之间的潜在关联。最后,研究了已鉴定的STs与其血清型和VAG谱之间的关系。
材料与方法
研究设计:尸检和抽样
预测FWM高于2%的禽群从家禽兽医处取样。鸡群来自挪威的不同地区和不同的杂交品种,尽管目前挪威的主要杂交品种是罗斯308(表1)1)。由一名家禽兽医对10只最近死亡或因动物福利原因而被实施安乐死的禽鸟/群进行尸检。在预定义的提交表格中注明肉眼病变的存在/不存在。在被检查的10只鸟中,有5只与大肠杆菌病相关的最典型的宏观病变被选择从脾脏、肝脏和Kravik等人先前描述的另一个器官进行细菌学检查。[12]。
细菌检验
每个样品被涂布到两个血琼脂(BA)板和一个心脏输注琼脂(HIA)板上,在37°C的CO中厌氧培养2根据Kravik等人所述的细菌诊断标准程序,分别在实验室和正常大气压下进行检测.[12]。孵育18-24小时后,检查所有样品是否存在大肠杆菌并对三种琼脂平板上菌落形态进行了描述。细菌的生长被分为稀疏、中等或丰富,以及纯度的水平大肠杆菌从1到4分:纯生长(1),几乎纯生长(2),主要生长大肠杆菌(3)和混合培养(4)。如果有几个菌落,则给2级肠球菌种虫害或普罗透斯sp .存在于琼脂上,与其他纯培养的大肠杆菌.三年级的定义是主导生长大肠杆菌,但结合稀疏到中等生长的任何一种Enterococcispp。普罗透斯一种或更少的不同细菌生长。混合培养,4级,是根据至少三种不同细菌的生长来定义的,其中大肠杆菌不是三个琼脂板上的主要细菌(附加文件1)。在细菌学检查中,至少有一个被证实大肠杆菌每个器官的分离物冷冻保存以备将来分析。
群诊断
大肠杆菌病的单独诊断是基于与大肠杆菌病败血症典型相关的病理病变的存在,并结合1-3级的细菌学检查。如果FWM高于1.5%,且该禽群的5只样本中至少有3只被单独诊断为大肠杆菌病,则定义为禽群诊断。
全基因组测序
在每一确诊大肠杆菌病的鸡群中,每群采集3-5只鸡,每只鸡采集1株分离菌进行WGS。每个分离物最好从肝脏中分离出来,并按前面所述提取DNA [12]。在挪威兽医研究所(NVI)制备了204株分离株的基因组DNA样本并进行了测序。克拉维克等人先前描述的另外15个分离株也包括在内。[12]。所有219株菌株均采用Nextera文库制备™DNA Flex文库制备(Illumina),并在Illumina MiSeq仪器上测序,得到300 bp的成对末端reads。本研究分析的序列数据在ENA数据库中公开,生物项目为PRJEB43441和PRJEB55163。参见附加文件1对于个人的加入号码。
计算机分析
全基因组序列组装与分型
Bifrost管道[13]用于初始质量控制和装配。该管道由读取质量控制、修剪、移除PhiX和组装组成。ARIBA [14版本2.14.6,根据Achtman方案确定序列类型(ST) [7]。具有新序列类型的分离株被上传到Enterobase进行ST定位[15]。使用SerotypeFinder进行血清型鉴定[16版本2.0.2。
Virulence-associated基因
使用VirulenceFinder 2.0.4版本进行VAGs检测分析。VirulenceFinder数据库通过添加致病菌毒力因子数据库中发现的已知apec相关基因(附加文件)进行了扩展2), (2,17,18,19]。完整的数据库包括629个毒力相关基因及其变异条目。
核心基因分析及系统发育分析
所有通过QC参数的分离株均根据核心基因进行系统发育分析。ALPPACA管道[20.版本1.0.0使用Prokka运行基因组注释[21版本1.14.6,随后使用Panaroo进行泛基因组分析[22在219个基因组中检测核心基因并进行比对。使用Snp-sites从比对中去除恒定位点[23版本2.5.1。Snp-dists [24],使用0.8.2版本计算与比对的成对SNP距离。最后,IQTree [25使用版本2.1.4生成系统发育树,使用ultraffast bootstrapping [26]有1000个重复,模型发现者加上[27]进行模型选择。
在两个最常见的STs (ST23和ST429)中,使用ALPPACA进行了单独的系统发育分析。ParSNP [28[1.6.1]版本生成核心基因组比对,然后使用Gubbins 3.2.0版本使用RaxML作为树生成器和GTRGAMMA模型检测重组区域。Maskrc-svg [29版本0.5随后用于掩盖重组区域。用SNP -sites去除固定位点,然后用SNP -dists两两计算SNP距离,用IQTree进行系统发育推断,同上。所有系统发育树均以R [30.版本4.0.2,使用版本3.0.4的ggtree包[31]。
结果
抽样和大肠杆菌病确认
2018年9月至2021年6月,对45只肉鸡和4只种鸡饲养群进行抽样调查,共获得49只肉鸡,FWM在1.53% ~ 12.6%之间。这些鸡群是不同的杂交品种,均小于14日龄,来自挪威不同地区,来自三个不同的孵化场。4个肉鸡群未被诊断为大肠杆菌病,因此被排除在进一步分析之外。总共有45只高FWM的鸡群被诊断为大肠杆菌病,并进一步纳入分析(表2)1)。
全基因组测序和计算机分析
质量控制
共确认219人大肠杆菌对分离株进行测序。一个大肠杆菌从每只鸟中选择3-5只鸟,从45个鸟群中取样(附加文件1)。基于multiQC和Quast报告的基因组序列初始质量控制结果显示,分离株GC含量在50.51 ~ 50.57%之间,contigs数为43 ~ 74,组装后的全基因组总长度为4.86 ~ 4.96 Mbp(参见附加文件中所有分离株的个体质量评分)1)。
MLST和血清分型
对来自45个禽群的219株APEC分离株进行了测序,并对其进行了ST和硅血清型鉴定。其中26个鸡群在同一群内的所有分离株中表现出相同的ST,而35个鸡群被鉴定出多达两种ST,因此在同一群内的至少三只鸟中发现相同的ST(表1)1和附加文件1)。在45个鸡群中,有10个在鸡群中发现了3个或更多的STs(表5)1)。
共鉴定出32种不同的STs,其中15种仅鉴定一次。本研究中最常见的STs依次为ST23、ST429、ST95、ST117、ST371、ST69和ST101(表1)2)。
计算机血清分型共显示38种不同的血清型。在单个ST中检测到一些血清型谱,而在多个ST中检测到其他血清型谱。在10种最常见的STs中,有5种STs具有几种血清型谱。最常见的血清型为O1:H7、O2/O50:H1、O2/O50:H5、O45:H19和O78:H42)。
系统发育分析
为了研究STs之间以及STs与血清型之间的关系,对所有219株分离株进行了核心基因分析(图2)1)。在219株分离株中,全基因组分析检测到14332个独特基因。其中,3303个被定义为核心基因,因为它们存在于至少95%的基因组中。模型查找器plus确定GTR + F + I + G4为最合适的模型。系统发育树分析结果显示,分离株根据其STs聚类,表明具有相同血清型谱的分离株可能存在遗传距离。O78:H4血清型分离株被鉴定为ST23和ST117, O2/O50:H1血清型被鉴定为ST429和ST135。此外,ST95和ST117还包含几种血清型谱:O1:H7和O2/O50:H5和O24:H4, O78:H4和O161:H4。

纳入研究的所有分离株的最大似然核心基因SNP树。大于或等于95的Bootstrap值表示为黑节点。核心基因树可视化了鉴定出的最常见序列类型(STs)之间的遗传关系以及与这些类型相关联的血清型谱。尖端和分支标签上的颜色分别代表血清型和STs,代表超过5个分离株。小费标签代表鸟群和鸟。
核心基因树中两个最大的簇分别由ST23和ST429代表。这两种STs都有一个不同的血清型谱,所有的鸡群,除了一个ST429的鸡群,根据杂交、采样日期和ST聚集在一起(图4)1、表1)。因此,这两宗st被怀疑代表了两次大肠杆菌病的爆发,因此对每宗st分别进行了系统发育分析(图2)。2和3.)。

最大似然核心基因组树显示了从17个鸡群(n = 81株)中鉴定为ST23的所有分离株的遗传关系。大于或等于95的Bootstrap值表示为黑节点。小费标签代表鸟群和鸟。

7个禽群(n = 33株)中鉴定为ST429的所有分离株的最大似然核心基因组树。大于或等于95的Bootstrap值表示为黑节点。小费标签代表鸟群和鸟。顶端的颜色代表混合型。进化枝A和进化枝C分别由来自3个禽群的分离株组成,均为杂交的Ross 308。进化枝B代表一个沙索群。
从17个禽群中共分离出81株被确认为ST23。所有ST23分离株均于2021年3月至2021年5月底从杂交罗斯308肉鸡群中采集1)。系统发育分析显示,ST23的平均基因组覆盖率为94.0%,SNP范围为0-33,SNP距离均值和中位数为15。ST23的个别分离株大部分按照单个禽群聚集,但也有分离株与来自不同禽群的分离株聚集(图3)2和表3.)。在一个鸡群中,除了两个鸡群外,其余鸡群的SNP平均距离都在10以下。一个群内SNP的范围从0 ~ 1(最小范围)到0 ~ 33(最高范围)不等(见表1)3.)。
在7个禽群的33个分离株中鉴定出ST429;2018年9月至2019年1月取样了6群杂交罗斯308,2019年8月取样了1群杂交萨索。系统发育分析显示,平均基因组覆盖率为95.8%,SNP范围为0 ~ 172,中位SNP距离为62。树状图显示,ST429分离株分为三个主要分支:ST429- a、ST429- b和ST429- c(图4)3.)。来自唯一的Sasso群的分离株聚集在ST429-B进化支中。分支ST429-A和ST429-C分离株来自不同地理位置、孵化场和亲本鸡群的罗斯308鸡群,但所有罗斯308肉鸡饲养群都分布在一个主要孵化场(图4)3.)。在ST429鉴定的单个鸡群中,除Sasso鸡群外,平均SNP距离均< 10个SNP, SNP范围从0 ~ 1到4 ~ 21不等(表4)3.)。
重新对ST429进行系统发育分析,只包括杂交的罗斯308群。该分析结果显示,平均基因组覆盖率为95.7%,SNP范围为0-76,中位SNP距离为59(附加文件)3.)。
Virulence-associated基因
从包含629个VAGs条目的数据库中,在本研究中至少一个APEC分离株中鉴定出112个VAGs变体(附加文件)1)。对ST23和ST429分离株以及包括所有其他分离株的第三组分别描述了VAGs的频率(图2)4)。

在所有ST23分离株中共鉴定出25个VAGs,而仅在部分ST23分离株中鉴定出5个VAGs。这些包括交易T,cba,美国中央情报局和cma分别在28%、17%、12%和17%的分离株中鉴定出。的迦得在94%的ST23分离株中鉴定出基因(图2)4)。所有ST429分离株均存在33个VAGs基因,部分分离株存在4个VAGs基因。的tsh,丰,增值税和迦得分别在61%、94%、12%和30%的ST429分离株中鉴定出基因(图2)4)。
在包含所有其他STs的第三组中,VAGs的存在具有更高的多样性。与ST23和ST429相似,第三组分离株均携带该基因csgf,ecp一个,ibeB,ibeC,iucC,坐D和的怪兽C.第三组中90%以上的分离株中存在其他几种VAGs,但也有少数分离株中不存在或仅存在VAGs(附加文件)1和图4)。铁载体受体基因fyu一个和irp2在所有ST23和ST429分离株中均存在,但在其他分离株中分别仅存在51%和52%。
讨论
关于APEC在挪威肉鸡生产中的作用的知识有限,在2014年北欧国家农场大肠杆菌病爆发高峰之后,我们认为有必要对APEC在挪威肉鸡生产中的流行病学进行系统研究。为了确定挪威肉鸡生产中的APEC类型,我们对高FWM鸡群进行了系统采样,并对这些鸡群中单个分离株的基因组进行了测序,以研究它们的STs、血清型谱、VAGs含量及其进化关系。
分离株的选择是根据2019年试点项目的结果进行的。该研究得出结论:(1)APEC在单个病禽中的多样性较低;(2)每群至少需要采样3只鸟,以确定一个禽群内APEC的多样性,并确定禽群水平上的主要致病APEC [12]。
在本研究中鉴定的32种不同的STs中,几乎50%只鉴定一次。在以前的研究中也观察到类似的趋势[32,33,34]。仅由一两个分离株代表的STs数量较多,影响了所报道的STs的高度多样性。目前尚不清楚这些单一ST菌株是真正的禽类病原体,对大肠杆菌病的病因学有重要意义,还是对致病性没有重要意义的零星发现。然而,这些报告强调,需要从一个畜群中对多个动物和多个分离株的WGS取样,以确定畜群中主要致病的APEC。
在45只病禽中,有35只被鉴定为主要的APEC型,其中ST23、ST117、ST371和ST429比其他病禽更容易单独引起疾病。后者表明这些STs在家禽中可能更具有致病性,而在鸡群中多个ST的混合感染中更常见的STs [35]。总共有10个鸡群表现出几种STs的组合,最常见的与其他STs组合的APEC类型是ST10, ST69, ST95和ST101。
值得注意的是,ST69和ST95在人类感染中经常被分离出来[5,36]。然而,由于它们存在于大多数混合感染中,因此很可能认为它们在家禽中是机会性的,而不是高致病性的。这得到了Kromann等人的支持,他们在非疫情情况下取样的健康家禽中确定ST95的患病率最高[37]。
然而,在讨论ST95时应谨慎,因为这种ST具有几种血清型谱,并且存在相同ST但具有不同毒力特性的变体[5,36,37]。我们的研究进一步展示了具有多种血清型谱的STs,如ST101,其中从三个鸡群中鉴定出O88:H8,从另一个鸡群中鉴定出O103:H21, ST117,从三个不同的鸡群中鉴定出O24:H4, O78:H4和O161:H4三种血清型。血清型O78:H4与ST23中发现的血清型相同。一般来说,亚太经合组织的分型应谨慎评估,正如核心基因分析所示,ST117和ST23在这些STs之间表现出很大的遗传距离,即使它们具有相同的血清型谱。在没有WGS数据的情况下,2014年ST117的两个明显的峰值[11]和2021年的ST23,可能有不同的起源,如果只进行血清分型,则被认为是相同的APEC,但如果进行MLST,则被认为是两个不同的APEC类型。另一方面,通过几种血清型谱确定的序列类型表明,两种分型方法的结合可以更好地区分APEC分离株(以便在爆发情况下快速诊断)[35]。
尽管亚太经合组织被认为是一种多样的病原体,但报告中最常见的STs之间的差异较小[12,33,35,38,39,40]。然而,某些STs的高流行率可能是由于大肠杆菌病在有限的时间内从单一来源小规模暴发,可能来自生产金字塔的高层[41]。
系统发育分析如今被认为是评估可能来自单一来源的分离株之间亲缘关系的金标准。因此,本研究给出了具有相同ST.的鸡群内部和鸡群之间的平均snp距离,该结果对于评估和了解未来农场间爆发的大肠杆菌病有价值。据我们所知,对于APEC疫情中可接受的snp数量的定义尚无共识[9,10,12]。突变率、病原体可能遇到的个体数量、病原体压力和源污染持续时间等因素都可能影响爆发期间出现的snp数量[9]。因此,在考虑暴发分离株之间的SNP距离时,应考虑家禽生产的金字塔结构以及病原体在引起肉鸡疾病之前在潜在的垂直传播线上将遇到的个体和代数。此外,在不了解已用于分析的基因组比例的情况下,不应该单独评估SNP距离。因此,病原体和种群元数据以及基因组覆盖率在了解分离物是否属于疫情方面发挥的作用与单核苷酸多态性距离一样大[9,10,12]。
总共有112个VAGs在我们的APEC分离物中至少被鉴定出一次。这与Apostolakos等人最近的报告相吻合.在他们的研究中发现了113个VAGs [32]。然而,在本研究中确定的两个主要STs (ST23和ST429)中,除了少数例外,存在较少的多样性。这是预料之中的,因为这些STs内的分离株被认为是同一次暴发的一部分,因此可能是克隆的。在ST23中变异的基因为达叻,cba,美国中央情报局和cma,并在ST429内增值税和迦得。已知这些基因大多数是质粒编码的,因此在一次疫情中不同分离株之间的差异可能比染色体编码的基因更频繁。此外,fyuA和irp2在所有爆发分离株中都有,但仅在大约一半的非爆发分离株中有。这两种VAGs都属于耶尔希菌控制子,该操纵子负责铁的获取,在禽大肠杆菌病的发病机制中具有重要意义[1]。
另一方面,在本研究的所有分离株中都鉴定出一些VAGs,因此可能被认为对APEC的毒力很重要。然而,如果没有一个真正的、非致病的对照组进行比较,就无法根据这些数据得出有效的结论,因为这些VAGs可能在所有禽类中都被鉴定出来大肠杆菌不考虑致病性的分离物。Johnson等人最近的一项研究[35]提示有两个与APEC质粒相关的保守VAGs,hlyF和施行了作为潜在的标记增加毒力潜力与其他遗传特征相结合。在本研究中,hlyF和施行了在所有ST23和ST429分离株中,以及89%和96%的其他STs分离株中分别鉴定出该基因。
在这项研究中,根据所鉴定的ST的流行程度及其在畜群内和农场间单独或联合造成高FWM的能力,评估了APEC菌株的毒力。此外,我们还深入了解了爆发菌株的相关性,并在我们的研究中提出了与两种爆发菌株ST23和ST429相关的最普遍的VAGs。然而,亚太经合组织的压力来自多方面大肠杆菌对于未来,追踪单一ST爆发,以确定是否相同的ST再次出现更容易引起整个农场的爆发,或者是否其他ST(在本研究中显得不那么重要)可能是未来爆发的原因,这将是很有趣的。在疫情爆发的情况下,更好地确定病原体在农场之间的传播途径将是一件有趣的事情。建议建立一个共同数据库,以控制和预防亚太经合组织的爆发[42]。对于未来,作者支持建立这样一个数据库,包括定义良好的元数据以及可比较的抽样和诊断方法。这样的数据库可以帮助鉴定通过肉鸡金字塔的病原体传播途径。此外,将系统取样的爆发菌株的VAGs与适当的对照组进行比较,将有助于进一步揭示个体APEC的毒力潜力。然而,界定亚太经合组织控制的重要性应进一步讨论,作为共同的大肠杆菌可能有潜力成为亚太经合组织,但其背后的病理生理机制仍未得到很好的界定[38]。
总之,本研究显示了2018-2021年挪威当地大肠杆菌病败血症爆发中发现的APEC类型的存在和分布。此外,它还确定了大肠杆菌病引起的高FWM峰是如何由单一的、独特的ST引起的。系统发育分析可以深入了解属于同一ST的分离株之间的相关性,也可以了解不同STs和血清型之间的相关性,确定需要结合分型方法来更好地区分APEC类型。这项研究还强调了使用WGS作为监测和确定未来大肠杆菌病暴发的诊断工具的价值。
参考文献
Nolan LK, Vaillancourt J-P, Barbieri NL, Logue CM(2020)大肠杆菌病。1 . Swayne DE, Boulianne M, Logue CM, McDougald LR, Nair V, Suarez DL, Wit S, Grimes T, Johnson D, Kromm M, Prajitno TY, Rubinoff I, Zavala G(主编)家禽疾病。威利·布莱克威尔,纽约
李建军,李建军,李建军,李建军(2005)禽致病性病毒毒力相关基因的快速检测大肠杆菌通过多重聚合酶链反应。鸟类疾病49:269-273
罗德奎,刘建军,刘建军(2005)亚太经合组织病理型的研究。箴言36:241-256
杨建军,杨建军,杨建军,杨建军。(2008)禽流感病原生物学的研究进展大肠杆菌毒力作为快速诊断工具。[J]中华临床微生物学杂志,46:39 - 39
Mehat JW, van Vliet AHM, La Ragione RM(2021)禽致病性大肠杆菌(APEC)病型由多个不同的、独立的基因型组成。鸟类病理学50:402-416
克莱蒙特O, christensen JK, Denamur E, Gordon DM(2013)克莱蒙特大肠杆菌重新探讨了类群分型方法:提高特异性和检测新的类群。环境微生物Rep 5:58-65
wwith T, Falush D, Lan R, Colles F, Mensa P, Wieler LH, Karch H, Reeves PR, Maiden MC, Ochman H, Achtman M (2006)大肠杆菌进化的观点。微生物学报,60:1136-1151
Lozica L, Repar J, Gottstein Ž(2021)自体疫苗应用对猪流感病毒序列类型和毒力谱影响的纵向研究大肠杆菌在肉鸡种鸡群中。兽医微生物学报259:109159-109159
Duval A, Opatowski L, Brisse S(2022)是一样的吗?定义针对个别疫情定制的基因组流行病学阈值。bioRxiv。https://doi.org/10.1101/2022.02.15.480545
陈建军,陈建军,李建军,等(2018)基于单核苷酸多态性的细菌全基因组分型和流行病学分析。临床微生物感染24:350-354
Ronco T, Stegger M, Olsen RH, Sekse C, Nordstoga AB, Pohjanvirta T, Lilje B, Lyhs U, Andersen PS, Pedersen K(2017)禽致病性传播大肠杆菌ST117 O78:北欧肉鸡生产中的H4。BMC Genomics 18:13
Kravik IH, Kaspersen H, Sjurseth SK, Jonsson M, David B, Aspholm M, Sekse C(2022)禽致病性高序列相似性大肠杆菌大肠杆菌病暴发期间从禽类个体和肉鸡群中分离出的菌株。兽医微生物学报267:109378
挪威兽医研究所/Bifrost:在工作顺序中发现的特定基因,https://github.com/NorwegianVeterinaryInstitute/Bifrost
李建军,张建军,张建军,张建军,张建军,张建军,张建军(2017)抗微生物药物耐药性基因分型研究进展。微生物基因组3:e000131
周忠,Alikhan N-F, Mohamed K, Fan Y, Achtman M (2020) EnteroBase用户指南(含案例研究)沙门氏菌传输,鼠疫杆菌发展史,大肠的核心基因的多样性。基因组Res 30:138-152
李建平,李建平,李建平,等(2015)血清型血清型的研究进展大肠杆菌利用全基因组测序数据分离。[J]中华临床微生物学杂志53:24 - 24
致病菌的毒力因子;埃希氏杆菌属,http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi
Kathayat D, Lokesh D, Ranjit S, Rajashekara G(2021)禽致病性大肠杆菌(亚太经合组织):概述毒力和发病因素,人畜共患的可能性,和控制策略。病原体10:467
Newman DM, Barbieri NL, de Oliveira AL, Willis D, Nolan LK, Logue CM(2021)禽致病性表征大肠杆菌(APEC)从大肠杆菌病病例,2018年。PeerJ 9: e11025
挪威兽医研究所,ALPPACA,第一次完整发布,(https://zenodo.org/record/6323152#.YwTGIdgzbcshttps://zenodo.org/record/6323152#.YwTGIdgzbcs)
Seemann T (2014) Prokka:快速原核基因组注释。生物信息学30:2068 - 2069
Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, Gladstone RA, Lo S, Beaudoin C, Floto RA, Frost SDW, Corander J, Bentley SD, Parkhill J(2020)利用Panaroo通道制备纯化的原核泛基因组。基因组生物学21:180
Page AJ, Taylor B, Delaney AJ, Soares J, Seemann T, Keane JA, Harris SR (2016) SNP-sites:多fasta比对的快速高效提取。微生物基因组2:e000056
Github, seemann, snp-dists,https://github.com/tseemann/snp-dists
陈建军,陈建军,陈建军,陈建军,陈建军,陈建军,陈建军,陈建军。(2020)IQ-TREE 2:基因组时代系统发育推断的新模型和有效方法。摩尔生物进化37:1530-1534
黄洪涛,陈志强,陈志强,陈志强(2017)一种基于自举法的超快速自举逼近算法。生物学报,35:518-522
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder:用于准确系统发育估计的快速模型选择。Nat Methods 14:58 - 589
Treangen TJ, Ondov BD, Koren S, Phillippy AM(2014)用于快速核心基因组比对和数千种内微生物基因组可视化的Harvest套件。基因组生物学15:524
Github, kwongj, maskrc-svg,https://github.com/kwongj/maskrc-svg
统计计算的R项目,https://www.R-project.org/
于刚(2020)基于ggtree的树状结构数据可视化。生物信息学[j] .中国生物信息学杂志69:96
Apostolakos I, Laconi A, Mughini-Gras L, Yapicier ÖŞ, Piccirillo A(2021)肉鸡大肠杆菌病的发生及其与禽致病性的关系大肠杆菌(APEC)群体结构与分子特征。《前沿兽医科学》8:737720
Cummins ML, Reid CJ, Roy Chowdhury P, Bushell RN, Esbert N, Tivendale KA, Noormohammadi AH, Islam S, Marenda MS, Browning GF, Markham PF, Djordjevic SP(2019)澳大利亚禽类致病性全基因组序列分析大肠杆菌它们携带1类整合酶基因。微生物基因组5:e000250
Delannoy S, Schouler C, Souillard R, Yousfi L, Le Devendec L, Lucas C, Bougeard S, Keita A, Fach P, Galliot P, Balaine L, Puterflam J, Kempf I (2020)大肠杆菌从法国日龄肉鸡中分离的菌株,其环境和80个鸡群的大肠杆菌病病变。兽医微生物学报252:108923
Johnson TJ, Miller EA, Flores-Figueroa C, Munoz-Aguayo J, Cardona C, Fransen K, Lighty M, Gonder E, Nezworski J, Haag A, Behl M, Kromm M, Wileman B, Studniski M, Singer RS(2022)禽致病性定义的改进大肠杆菌(APEC)通过纳入高危克隆群体的病理类型。家禽科学101:102009
Jørgensen SL, Stegger M, Kudirkiene E, Lilje B, Poulsen LL, Ronco T, Pires Dos Santos T, Kiil K, Bisgaard M, Pedersen K, Nolan LK, Price LB, Olsen RH, Andersen PS, Christensen H(2019)鸟类与人类的种群重叠大肠杆菌属于95式序列。mSphere 4: e00333-e418
Kromann S, Baig S, Stegger M, Olsen RH, Bojesen AM, Jensen HE, Thøfner I(2022)肉鸡种鸡群及其后代背景病变的纵向研究和基因组特征大肠杆菌.Vet Res 53:52
Mageiros L, m
王晓明,王晓明,王晓明,等(2019)ESBL/ pampc的垂直传播大肠杆菌有限的金字塔家禽生产。兽医微生物学报231:100-106
李建军,李建军,李建军,李建军,李建军,李建军。(2020)中国水稻基因组学研究进展大肠杆菌从捷克共和国的病鸡中分离出的菌株。BMC兽医Res 16:189
李建军,李建军,李建军,李建军,李建军,李建军,李建军(2017)疾病传播的纵向研究大肠杆菌从肉鸡饲养员到肉鸡。兽医微生物,2007:13 - 18
Christensen H, Bachmeier J, Bisgaard M(2021)禽致病性防控新策略大肠杆菌(亚太经合组织)。禽病50:370-381
致谢
本研究由挪威研究理事会农业和食品工业研究基金(NFR项目280385)资助。生物信息学分析是根据挪威国家高性能计算和数据存储基础设施uninet Sigma2提供的资源进行的。作者要感谢贡献的兽医Astrid s . yland Grødem, Lena Hollund Surdal, Mona Nordmark (Den Stolte Hane), Aslak Fitjar Oltedal, Torill Kjensmo, Marielle Wigaard, skjul Arne Hansen, Pia k . teles, Elisabeth Kverneland (Nortura), Tove Elin Sande, Sigrid Gosse, Morten h . glien, Miriam Garner (Norsk Kylling)和Kate Norum (Ytterøy Kylling)。特别感谢细菌学实验室的Bjarne Bergsjø, Bereket S. Tesfamichael, Basma Asal和Fiona Valerie Franklin-Alming,以及挪威兽医研究所全基因组测序的catherine Arnason Bøe和SEQ-TECH在实验室的持续帮助和支持。
作者信息
作者及单位
贡献
SKS, BD, MA, CS对研究进行了构思和设计。IHK, SKS, CS组织了取样和实验室分析。IHK, HK分析了数据。香港的统计和生物信息学方法得到KDR的协助。IHK, HK, SKS, BD, CS解释数据。IHK准备了手稿的初稿。HK, SKS, KDR, BD, MA, CS阅读,参与并批准最终稿件。所有作者都阅读并批准了最终的手稿。所有作者都阅读并批准了最终的手稿。
相应的作者
道德声明
相互竞争的利益
作者宣称他们没有竞争利益。
额外的信息
执行编辑:Pauline Ezanno
出版商的注意
伟德体育在线b施普林格《自然》杂志对已出版的地图和机构的管辖权要求保持中立。
补充信息
附加文件1。与本研究中测序的每个分离物相关的元数据包括群和采样数据、细菌学分级、测序质量措施、鉴定的毒力相关基因和ENA的加入号。
1样本ID解释了从哪个鸟群(第一个数字)和哪个鸟(最后一个数字)中取样分离物。2根据细菌培养物在三个琼脂板上生长的纯度进行分级,在常压、CO2室或厌氧条件下培养过夜:1级=纯生长,2级=很少菌落肠球菌种虫害或普罗透斯sp .存在于琼脂上,与其他纯培养的大肠杆菌。3级=主导生长大肠杆菌稀疏到中等生长的Enterococcispp,普罗透斯一种或更少的不同细菌的生长,4级=混合培养:至少有三种不同的细菌,其中大肠杆菌并不是主要的细菌。3.在单个鸟的尸检中存在与大肠杆菌病(大肠杆菌血症)相关的病理病变,≥2个病变为1,<2个病变为0。4如果有大肠杆菌血症的病变,每只鸟都被诊断为大肠杆菌病(1),个体样本的细菌学检查等级为1-3。
附加文件2。上传至VirulenceFinder的基因扩展数据库中用于本研究中毒力相关基因鉴定的基因列表。
表中包括基因的名称、基因的简短描述以及从哪个数据库(VirulenceFinder或VFDB)中鉴定出的基因。
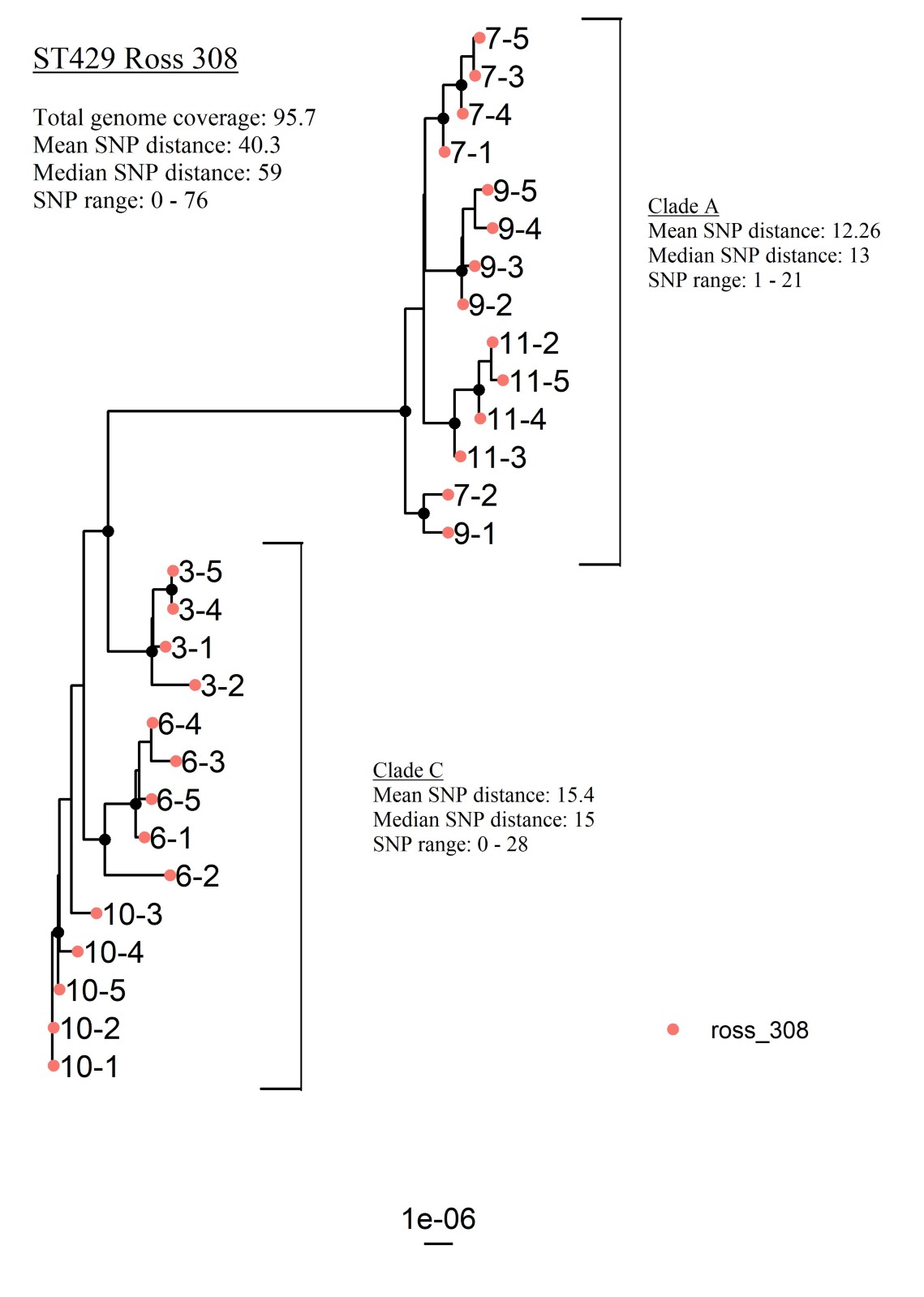
附加文件3。最大似然核心基因组树显示了所有罗斯308鸡群(n = 28)中鉴定为ST429的所有分离株的遗传关系,不包括来自一个萨索鸡群的分离株。
{kind=link}
大于或等于95的Bootstrap值表示为黑节点。小费标签代表鸟群和鸟。进化枝A和进化枝C分别由来自3个禽群的分离株组成,均为杂交的Ross 308。
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议,该协议允许以任何媒介或格式使用、共享、改编、分发和复制,只要您适当地注明原作者和来源,提供知识共享许可协议的链接,并注明是否进行了更改。本文中的图像或其他第三方材料包含在文章的知识共享许可协议中,除非在材料的署名中另有说明。如果材料未包含在文章的知识共享许可中,并且您的预期用途不被法律法规允许或超过允许的用途,您将需要直接获得版权所有者的许可。如欲查阅本许可证副本,请浏览http://creativecommons.org/licenses/by/4.0/.创作共用公共领域免责声明(http://creativecommons.org/publicdomain/zero/1.0/)适用于本文中提供的数据,除非在数据的信用额度中另有说明。
关于本文
引用本文
克拉维克,i.h.,卡斯珀森,h.h.,舒尔塞斯,S.K.et al。的分子流行病学研究大肠杆菌在患有大肠杆菌病的雏鸡中,在整个农场发现了两次可能的暴发。兽医Res54, 10(2023)。https://doi.org/10.1186/s13567-023-01140-6
收到了:
接受:
发表:
DOI:https://doi.org/10.1186/s13567-023-01140-6
关键字
- 禽致病性大肠杆菌(亚太经合组织)
- 大肠杆菌病
- 家禽
- 全基因组测序
- 毒力相关基因
- 爆发
- 系统抽样