摘要
生物体液的代谢组学时间过程分析与临床诊断高度相关。然而,许多抽样方法受到未知样本大小的影响,通常被称为大小效应。这阻碍了生物标记物的绝对定量。最近,通过利用已知的药代动力学信息或通过统计手段,开发了几种数学上的获取后归一化方法来克服这些问题。在这里,我们提出了一种改进的归一化方法MIX,它结合了两种方法的优点。它将两个归一化项结合在一起,一个基于药代动力学模型(PKM),另一个代表一种流行的统计方法,概率商归一化(PQN),在一个模型中。为了测试MIX的性能,我们生成了与真实手指汗液代谢组测量值非常相似的合成数据。我们表明MIX归一化成功地解决了单个策略的关键弱点:它(i)降低了与PKM过拟合的风险,(ii)与PQN相反,它允许计算样本体积。最后,我们通过使用真实手指汗液和血浆代谢组数据验证MIX,并证明MIX可以更好、更稳健地校正尺寸效应。总之,MIX方法提高了手指汗液和其他生物体液中生物标志物定量检测的可靠性和鲁棒性,为从代谢组学时间过程数据中发现生物标志物和生成假设铺平了道路。
介绍
近年来,汗液代谢组的分析越来越受到几个研究领域的关注[1,2,3.]。例如,汗液一直是法医科学家关注的焦点,因为它有可能分析已发现的指纹的代谢组学特征(例如,在犯罪现场)[4]。此外,药物测试可以很容易地在汗液样本上进行。这种方法的一个优点是不仅可以识别已经非法的物质,而且还可以识别它们的代谢降解产物,从而使科学家能够区分药物消费和仅仅接触[1]。汗液代谢组学的另一个应用是个性化医疗诊断,其重点是识别身体的代谢状态,并试图根据汗液中的生物标志物信息优化营养和治疗[5,6,7]。
汗液代谢组学提供了几个技术优势。首先,汗液是生物分子的丰富来源,因此为生物标志物的发现提供了巨大的潜力[8,9]。其次,与其他生物液体(如血液或尿液)的取样相比,汗液取样比较容易。此外,它是非侵入性的,原则上可以快速重复。
已经发展了几种抽样方法[2,3.,9,10]。然而,它们中的大多数都以非常相似的方式工作:在皮肤表面涂上一层吸水材料,在一段(短)时间内收集汗水。随后从该材料中提取汗液代谢物并进行分析[3.,10]。然而,不同的方法在是否以及如何引起出汗方面有所不同。有些方法通过体育锻炼诱导出汗增加[9]或化学刺激[2],而在其他研究中,没有进行排汗诱导,自然排汗率足以进行代谢组学分析[3.,11]。
无论采用何种精确的抽样方法,上述大多数研究都有一个主要缺点。汗液通量变化很大,不仅取决于个体间差异,还取决于身体位置、温度、湿度、运动和其他可能在一天内多次变化的因素[12,13]。例如,即使保守估计汗液通量的可变性,文本\ (q_ \{汗}\),在指尖0.05至1毫克厘米之间\ (^ {2} \)最小值\ (^ {1} \)需要被考虑[13,14,15,16]。这对比较或定量研究来说是一个重大挑战,许多人已经认识到这一点,例如。[1,4,8,17,18,19],但只有少数人主动接近,最引人注目的是[9]。
关键问题在于人们通常对真正的代谢物浓度感兴趣,在{\ (\ mathbf C {} \ \ mathbb R} ^ {n_{\文本{代谢物}}}\)的,\ ({n_{\文本{代谢物}}}\)代谢物,这是由一个未知的和随时间变化的汗液通量模糊。因此,所测代谢物的强度与\ (\ mathbf C {} \)但是对于代谢物质量矢量,\ (\ widetilde {\ mathbf {M}}在{\ mathbb R} \ ^ {n_{\文本{代谢物}}}\),
在这里文本\(现代\{样本}\)和\ \(τ\)分别表示被采样皮肤的表面积和采集一个样本所需的时间。我们强调,在整个手稿中,代谢物的质量被定义为测量样品中代谢物的测量丰度,而不是摩尔质量或质量电荷比。此外,我们承认,如果没有校准曲线,所测量的丰度具有任意的峰面积单位,因此严格地既不是绝对质量也不是绝对浓度。将测量强度换算成质量单位的比例常数由校准曲线确定。适当的校准曲线在这里没有进一步讨论,但假设是线性的,并且在适用时可用。
代谢浓度变化发生在两位数的分钟到小时的范围内,而采样时间通常是低个位数的分钟,因此可以假设\ (\ mathbf C {} \)在集成时间内变化很小\ \(τ\)[20.]。因此,(1)简化为
取样时汗液量未知
问题是:给定\ (\ widetilde {\ mathbf {M}} \),我们如何计算\ (\ mathbf C {} \)如果我们不知道\ \ (V)?
从体积未知的小生物样本中计算绝对代谢物浓度的需求并非汗液代谢组学所独有,而是在非靶向代谢组学中已知的。这个问题通常被称为尺寸效应[21]。为了与之前关于该主题的出版物保持一致,我们将在本出版物中使用术语“大小效应”。我们强调,在这种情况下,它特指由于样品体积和/或稀释度的变化而引起的测量丰度的感知差异,而不是每个样品的不同测量次数的影响,也称为样本大小效应[22]。
已经制定了三个策略来解决规模效应:
直接测量汗液量。测量\ \ (V),例如通过微流体[9,23,24,是解决问题最直接的方法。2),通常非常准确,所需的最小体积在\ (5 \ \ sim)到100年\ \ upmu \ ()L (9,23,24]。然而,在汗液取样的情况下,可能需要相当长的时间,较大的取样面积,或增加(即诱导)出汗来收集足够的汗液以进行稳健的体积量化。另一种选择是通过配对标准进行体积估计[25],然而,这种方法增加了样品制备的复杂性。任何一种选择都会阻碍快速简便的样品采集和分析。
间接汗量计算。如果已知目标代谢物浓度的化学动力学,则通过将测量的质量矢量拟合到式,可以同时确定动力学参数和每个时间点的汗液量。2.最近,我们使用这一策略不仅在计算上将nL中的样本体积分解为个位数\ \ upmu \ ()l -范围,但也能准确量化咖啡因摄入后的个性化代谢反应模式[20.]。虽然在反应动力学知识的基础上确定个体差异是可行的,但当可用的先验信息太少时,这种方法很快就会变得不受约束。因此,它不适合发现未知的反应动力学。此外,该方法需要几个采样时间点,以便对不同代谢物的动力学进行建模,从而降低了采样的简单性。
统计规范化。使用这种方法的目的是通过与样本量成比例缩放的标记物的表观质量对质量矢量进行归一化,以便该比率(至少近似地)与样本量无关。针对非靶向代谢组学开发了各种策略;例如,按总测量信号归一化[26],以及基于奇异值分解的归一化[27]。然而,性能最好的方法之一——概率商归一化(PQN)——简单地假设两个表观质量向量之比的中位数与样本体积成正比[21,28,29,30.]。尽管PQN不允许计算样本体积本身,它使人们能够评估不同的变化[28]。
在本研究中,我们探讨了三种不同的归一化方法对合成数据的性能。我们说明了两种先前发表的方法的缺点,分别只关注目标或非目标代谢物。第三种规范化方法是将这两种策略组合在一个MIX模型中。我们表明MIX显著优于之前的归一化方法。为了验证结果,我们使用MIX来表征手指汗液中测量的咖啡因代谢以及血浆中测量的苯海拉明代谢。
理论
概率商归一化
定义。概率商归一化(PQN)假设,对于大量非目标代谢物,两个样本(例如,两个测量时间点)之间的中位数代谢物浓度变化倍数,\ (t_r \)和\ (t_s \))近似为1,
因此,折叠变化从\ (\ widetilde {\ mathbf {M}} \)而不是\ (\ mathbf C {} \)与的比率成正比吗\ \ (V),
与
以尽量减少实验误差的影响
常取代专用样品中的\ (\ widetilde {M} _j (t_s) \)在方程的分母中。3 c[28]。因此,PQN的归一化商计算为
文本\(问^ \ {PQN} \)是一个相对度量,分布在1附近。类似于Eq。3 b,我们定义其与(汗液)体积的关系{PQN} \ \ (V ^ \文本)作为
在哪里\ (V ^文本{ref}} {\ \)表示未知的、与时间无关的参考(汗液)体积。请注意,仅使用实际数据{PQN} \(问^ \文本(t) \)值可以计算,但是{PQN} \ (V ^ \文本(t) \)以及\ (V ^文本{ref}} {\ \)仍然未知。
讨论。{ref} \ \ (M_j ^ \文本)可以根据底层数据进行不同的定义。然而,参考文献的选择通常对PQN的结果并不重要[28]。由于没有可用的对照或空白测量,并且代谢物的丰度可以在几个数量级的范围内,在本研究中,我们使用代谢物的中位数参考文本\(问^ \ {PQN} \)计算。此外,PQN可能对缺失值敏感;然而,在本研究中,我们只关注(真实的和合成的)数据集,其中100%的值存在。
PQN的最大优点是不需要校准曲线和关于测量代谢物随时间变化的先验知识。此外,PQN与时间序列中测量的样本点数量无关。然而,它的主要缺点是,归一化商不是一个绝对的量化,只显示相对的变化。也就是说,它没有量化\ \ (V)如式所示。2直接使用绝对值,而不是标准化样本和时间点之间的相对丰度。另一个关键的假设是,汗液代谢物浓度在采样时间序列中平均需要保持恒定。鉴于对于健康人的汗液来说这是合理的假设[20.],人们在调查疾病状态时必须小心(例如,囊性纤维化,已知会改变汗液的成分[31])。
药代动力学正常化
定义。在药代动力学模型(PKM)中,我们假设我们至少知道药代动力学的功能依赖性,但不一定知道药代动力学的值k(药学)动力学参数\(\varvec{\uptheta}\in {\mathbb R}^k\)为\(2\le \ well \le {n_{\text{代谢物}}})代谢物。在不失一般性的前提下,我们(重新)排序\ (\ widetilde {\ mathbf {M}} \)这样第一个\ \)(\拼元素(在vector中收集)\ (\ widetilde {\ mathbf {M}} _ \魔法\))对应于已知药代动力学依赖的代谢物,而其余的\({n_{\text{代谢物}}}- \ well \)元素(在vector中收集)\(\ widdetilde {\mathbf {M}}_{\ell +}\))对应于动力学未知的代谢物。那么情商。2采取的形式是
有物理意义的界限;
{11} \ (V ^ \文本(t) \)以及\ (\ varvec {\ uptheta} \)的参数拟合可以得到\(\ widdetilde {\mathbf {M}}_\ well ^\text {PKM}(t)\).注意,这不仅允许计算的绝对值\ (\ mathbf {C} _{11} \魔法^ \文本(t; \ varvec {\ uptheta}) \)But - with{11} \ (V ^ \文本(t) \)-所有其他浓度也通过\ (\ mathbf {C} _ {\ l形+}(t) = \ widetilde {\ mathbf {M}} _ {\ l形+}(t) / V ^ {11} (t) \ \文本).
作为{11} \ (V ^ \文本(t_i) \)可能每个时间步都不一样\ (t_i \),我们需要知道至少两种代谢物的(药物)动力学;否则,参数数大于数据点数。
讨论。该方法的最大优点是可以隐式估计的绝对值\ \ (V)不需要直接测量。因此,汗液体积可以变得比体积法所需的最小体积更小,并且更短的采样时间也成为可能。这种方法的一个缺点是,它只有在具有相关药理学参数的先验知识(即,感兴趣的代谢物的摄入剂量,分布体积,标本的体重,预期动力学常数的范围)时才可行,这限制了至少两种代谢物及其药代动力学已知的研究方法。此外,为了稳健性地拟合方程系统,需要感兴趣的代谢物的校准曲线和时间序列中足够多的样品。在先前进行的敏感性分析中,当每个测量时间序列的样本数量从15个时间点增加到20个时间点时,观察到拟合质量的增加[20.]。
混合正常化
定义。混合归一化模型(MIX)是PQN和PKM的结合。它旨在通过其PQN术语纳入非靶向代谢组学的强大统计数据以及绝对估计\ \ (V)通过它的PKM项。
通过对两个方程的优化得到MIX的最优参数;
和
其中附加的转换T(PKM和PQN项)和缩放Z(PQN术语)可用于解释随机和系统误差(参见Hyperparameters”),{混合}\ (V ^ \文本(t) \)和\ (\ varvec {\ uptheta} \)被限制在物理意义上的界限之间,
例如:\ \ (V)可由式计算。2 b最小流汗率和最大流汗率。
讨论。我们假设MIX模型可以结合PQN和PKM归一化模型的优点。此外,我们认为MIX继承了PQN的统计鲁棒性,同时估计了PKM拟合的绝对值。使用PKM或MIX进行规范化需要几个先决条件。然而,如果满足这些要求,使用MIX而不是PKM所提高的归一化的好处通常不会带来额外的代价,因为在许多代谢组学研究中,靶向代谢物和非靶向代谢物是联合测量的,因此,MIX所需的所有额外数据都已经可用。
方法
实现
PKM和MIX(其中可以对任意数量的独立代谢物动力学进行建模)的通用版本被实现为Python类。作为输入,它需要用于动力学建模的代谢物的数量(\ \)(\拼)、时间点矢量以及测量的质量数据(\ (\ widetilde {\ mathbf {M}} \)(以时间点为行,代谢物为列的矩阵)。MIX还需要一个文本\ (\ mathbf {Q} ^ \ {PQN} =[问^ \文本{PQN} (t_1)……,Q^\text {PQN}(t_{n_{\text {time points}}})]^\text {T}\)载体(用PQN法从所有代谢物中计算,\ ({n_{\文本{代谢物}}}\))表示时间序列的所有时间点。优化后(执行)self.optimize_monte_carlo,这是SciPy的包装器optimize.curve_fit[32])动力学常数和汗液体积通过最小化公式中列出的函数来优化测量数据。9 b和9 c分别为PKM和MIX:
在哪里
\(\text {Var}(\mathbf {V})\)的方差\ (\ mathbf {V} \)(这是估计的向量V在所有时间点上),T是一个变换函数,Z是缩放函数,然后呢l是损失函数。PKM和MIX的关键区别在于拟合\ \ (V)的相对丰度是由PQN计算得到的。MIX模型的一个重要的附加超参数是\λ(\ \)的误差残差加权\(\mathcal {L}^\text {PKM}\)和\(\mathcal {L}^\text {PQN}\).它的计算在“Hyperparameters”。如果\(\lambda = 1\), MIX模型再次简化为纯PKM模型。
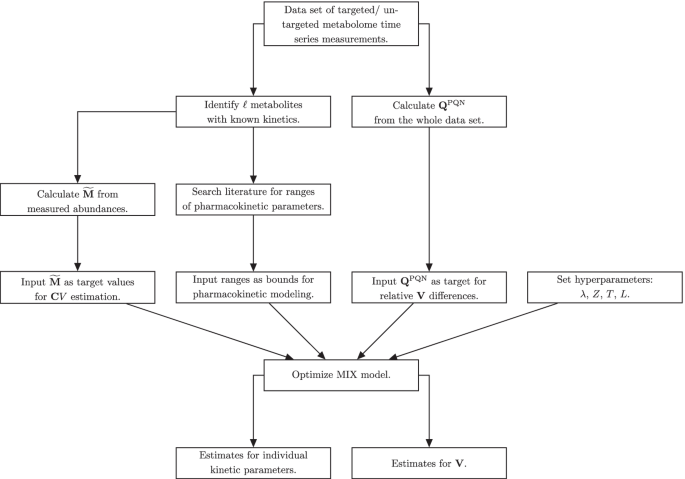
用于MIX规范化的数据处理流程图
总之,附加文件中给出了PKM和MIX模型之间差异的概述1:表S1和MIX归一化数据处理流程图如图1所示。1.
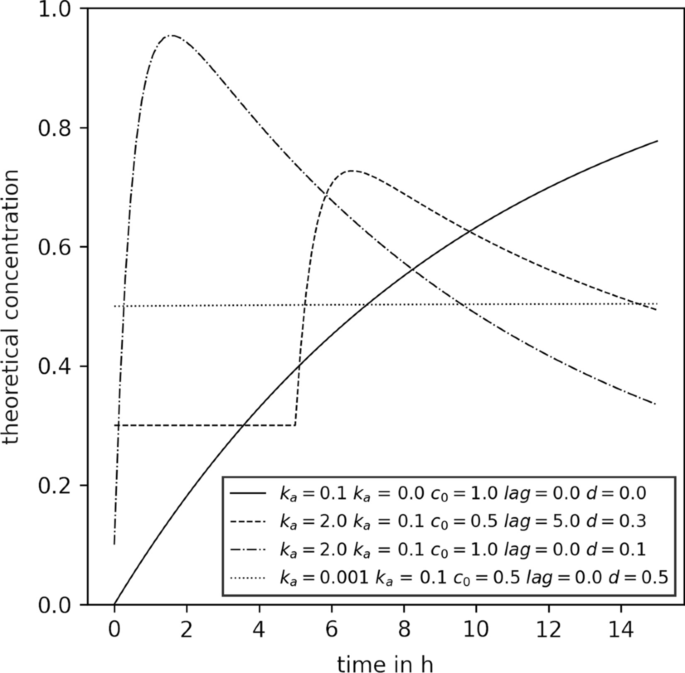
浓度时间序列的例子,可以用修正的贝特曼方程来建模。图例显示了用于创建各自曲线的动力学参数。所有参数都在动力学参数拟合的范围内
Hyperparameters
可以为PKM和MIX Python类设置几个超参数。
动能函数。首先,可以选择用于计算的动力学函数\ (\ mathbf C {} \).在本研究中,我们关注的是一个修正的贝特曼函数F(t),有5个动力学参数(\ (k_a ~ k_e, ~ c_0, ~滞后,~ d \)):
与
这个函数被设计得很灵活,能够代表几种不同的代谢物消耗和生产动力学,如图2所示。2.直观地说,\ (k_a \)和\ (k_e \)用单位h表示感兴趣的代谢物的吸收和消除的动力学常数\ (^ {1} \).\ (c_0 \)代谢产物的吸收总量除以单位摩尔L的分布体积\ (^ {1} \).此外,这些参数也是经典蝙蝠侠函数的一部分[33,我们在此介绍滞后和d.的滞后单位为h的项将函数沿x轴移动,直观地定义了感兴趣的代谢物吸收的开始时间点,而d单位mol L的项\ (^ {1} \)将函数沿y轴移动。
损失函数,l.l计算模型误差残差估计后的损失值(Eq。9).它可以通过设置self.set_loss_function要么cauchy_loss或max_cauchy_loss(或max_linear_loss).在这两种情况下,损失都是根据SciPy计算误差残差的柯西分布[32]。然而,不同之处在于cauchy_loss只使用绝对误差残差,而max_cauchy_loss使用相对和绝对误差残差的最大值(因此单词马克斯由其名称表示)。增加的原因是在先前的一项研究中取得了良好的表现[20.]。在这项研究中,我们使用了max_cauchy_lossPKM模型的损失函数和cauchy_loss用于MIX模型。的选择l与选择交织在一起T这一点在下一段中变得很清楚。
转换函数,T.T转换测量数据\ (\ widetilde {\ mathbf {M}} \)以及计算出来的\(\mathbf {Q}^\text {PQN}\),\ (\ mathbf V C {} \),\ (\ mathbf {V} \)在计算损失之前(Eq。9).两个不同的变换,没有一个和log10,可以在初始化时使用参数进行设置trans_fun.正如最初报道的[20.]没有对PKM进行转换(即:trans_fun = '没有'),
然而,对于MIX模型,执行对数变换(即:trans_fun = ' log10 '),
由于测量数据上的误差被认为是乘法[34]和汗量对数正态分布(附加文件1:图S1)。为了避免大小为0的浓度的问题,一个小的数字(即优化器精度的大小[32])。
在一项敏感性分析研究中,我们测试了MIX与不同的归一化质量l和T并得出超参数的组合cauchy_loss为l和log10为T执行最佳(附加文件1:图S2C, D)。这与文献一致,其中对数变换与PQN结合在汗液测量的尺寸效应归一化方面表现良好[35]。
尺度函数,Z.Z描述在上执行的缩放函数\(T(\mathbf {Q}^\text {PQN})\)和\ (T (\ mathbf {V}) \).执行缩放以纠正有噪声的数据(参见“结果”部分)。噪声对PQN的影响”)。可以设置两种策略scale_fun参数在MIX模型类初始化期间,标准或的意思是.在本研究中,所有MIX模型均采用标准尺度,即。
我们还实现了的意思是缩放取决于选择T与
优化策略。PKM和MIX模型的优化都是通过蒙特卡罗策略完成的,其中初始参数从其边界之间的均匀分布中随机采样。进行敏感性分析,我们之前表明这种方法优于单一拟合程序[20.]。在本研究中,模型拟合的蒙特卡罗重复数设置为100。
MIX损失项的加权。模型可以使用每个测量数据点的加权常数。在一项敏感性分析研究中,我们发现\λ(\ \)对标准化的质量不是至关重要的,只要它不是极端地偏向一边(即,\λ(\ \)接近0或1,附加文件1:图S2A, B)。因此,我们提出了一种方法,其中损失项由两个损失项的每个拟合数据点的数量加权,而不是由计算每个项时使用的代谢物的数量加权(附加文件)1:式S1)。对于这种方法,解为\λ(\ \)由式给出。13.
全模型和最小模型
在本研究中,我们区分了满模型和最小模型。对于完整模型,我们参考药代动力学归一化模型(PKM或MIX),其中给定数据集的所有代谢物用于药代动力学归一化。这意味着,例如,如果\({n_{\文本{代谢物}}}= 20\)所有20种代谢物都用改良的贝特曼函数建模,因此在式中。7一个和8,\(\ well = {n_{\text{代谢物}}}\)和\(\ widdetilde {\mathbf {M}}_{\ell +}\)是一个空向量。另一方面,最小模型是指只有少数已知的、受到更好约束的代谢物用动力学函数建模的模型。这意味着该信息用于PKM\(_{\文本{最小}}\)不改变(合成)代谢物的添加。因此,当的变化时,其拟合优度应在统计变异性内保持不变\ ({n_{\文本{代谢物}}}\).这种行为被用来验证模拟是否按预期工作,以及随机数生成是否存在偏差。另一方面,MIX\(_{\文本{最小}}\)模型仍然从的增加中获得信息\ ({n_{\文本{代谢物}}}\)因为该模型的PQN部分是用all来计算的\ ({n_{\文本{代谢物}}}\).因此,MIX的拟合优度指标的变化\(_{\文本{最小}}\)预计。我们强调,充分和最小模型的定义是具体到这个特定的研究。这里我们明确地设置\(\ well = 4\),这源于先前的工作,其中4种目标代谢物(咖啡因,副黄嘌呤,可可碱,茶碱)具有已知的动力学测量[20.]。
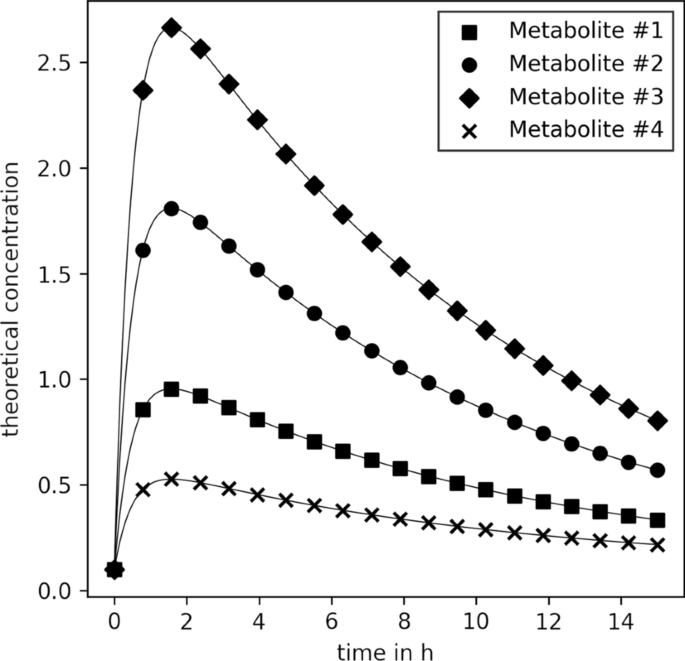
理论浓度\ (\ mathbf C {} \)为前四种代谢物的合成数据。用于计算的动力学参数列在附加文件中1表S2
合成数据创建
研究了三种不同类型的合成数据集。前两种类型的数据集(从动力学中取样,参见采样动力学"从均值和标准差中抽样"抽样均值和标准差”)测试归一化模型在极端情况下的行为(要么所有代谢物都可由药代动力学描述,要么所有代谢物完全随机)。最后,第三种类型的数据集(从真实数据中采样,参见“从真实数据中取样)的目的是尽可能地复制测量到的手指汗液数据。总而言之,规范化方法在所有三种类型数据集上的性能可以显示它们在具有不同数量可描述数据的不同情况下的行为。
在这三种情况下,数据的创建都是从一个简单的玩具模型开始的,该模型与手指汗液中咖啡因及其降解产物(副黄嘌呤、可可碱和茶碱)的浓度时间序列非常相似。20.]。附加文件中列出了相应的参数1表S2。利用它们,计算20个时间点(在等距离间隔的0 - 15h之间)代谢物#1至#4的浓度,图2。3.).随后,对新的合成代谢物浓度时间序列进行采样,并将其附加到玩具模型(即浓度向量)中,\ (\ mathbf} {C (t) \)).测试了三种不同的合成数据采样策略,下面几节将解释它们的具体细节。接下来,汗量(\ \ (V)的对数正态分布,截短于(\(0.05 \le V\le {4}\,\upmu \hbox {L}\))与我们在之前的刊物[20.],附加文件1:图S1。最后,一个实验误差(\ (\ varvec {\ upepsilon} \)),每个代谢物和时间点均服从正态分布,变异系数为20 \ % \ \ ()合成数据计算为
对于每个测试条件,生成100个合成数据重复,并拟合归一化模型。
采样动力学
在模拟v1中,数据是通过从均匀分布中采样新代谢物的动力学参数来生成的。分布受到与PKM和MIX模型拟合相同的边界约束:\ ((0, 0, 0, 0) ^ {T} \ \文本勒(k_a、k_e c_0,滞后,d) ^ {T} \ \文本勒(3)3、3、5、15日^ {T} \ \文本).然后根据修正的贝特曼函数(Eq.)计算合成代谢物的浓度时间序列。10).
抽样均值和标准差
代谢物浓度时间序列的均值和标准差由非目标的真实手指汗液数据计算(详见“真实手指汗液代谢组数据”)。两者的概率密度函数都可以用对数正态分布来描述(附加文件)1:图S3)。对于模拟v2的数据生成,每个添加的代谢物,从拟合分布中采样一个平均值和一个标准差,并将其用作另一个对数正态分布的输入,随后从中采样随机浓度时间序列。这导致合成浓度值的行为是随机的,因此,不能轻易地用我们的药代动力学模型来描述。
从真实数据中取样
为了更好地接近真实数据,在模拟v3中,浓度时间序列直接从非目标的真实手指汗液数据中采样(详细信息请参见“真实手指汗液代谢组数据”)。为此,非靶向代谢物\ (\ widetilde {\ mathbf {M}} \)用PQN对时间序列数据集进行归一化。由于该数据集中代谢物的数量相当大(\({n_{\文本{代谢物}}}= 3446\)),我们可以假设相对误差(或rRMSE,有关更多解释,请参见“综合数据模拟)是微不足道的。严格地说,结果值是浓度的分数。然而,这并不影响结果,因为这些值无论如何都被认为是非目标的(即,不存在校准曲线),因此是相对的。因此,PQ归一化数据集可以作为浓度时间序列采样的基真值。随后,对原始地面真值数据的一个子集进行采样以生成合成数据。
噪声数据的采样
研究了背景(即噪声)信号对系统性能的影响\(\mathbf {Q}^\text {PQN}\)(以及其缩放和变换的变体)。为了模拟这样的环境,我们使用了从真实数据中采样的数据(参见“从真实数据中取样),然后申请\ \ (V)只有一小部分\ (\ mathbf C {} \)向量,
的元素个数给出了噪声分数\ (\ widetilde {\ mathbf {M}} \)和\ (\ widetilde {\ mathbf {M}} _n”\)向量,
在下标n在\(\ widdetilde {\mathbf {M}}_n, \mathbf {C}_n,\)和\ (fn \)表示它们是噪声的一部分。
对20个等距噪声分量进行了仿真\(0 \ \ f_n \le 0.95\)与\({n_{\文本{代谢物}}}= 100\)和\(n_{\text{时间点}}= 20\)对于100个重复。均值和标准的误差残差\(\mathbf {Q}^\text {PQN}\)计算如下
与Z定义为式。12 b和
与Z定义为式。12个一个.对于这两种情况T定义为对数(Eq。11 b).我们指出乘法与\(\text {Std}(T(\mathbf {V}))\)对于标准标度误差来说,使结果具有可比性是很重要的,否则无论标度的性能如何,误差都会偏向于标度标准差较小的方法。
归一化模型优化
通过测量值拟合动力学对汗液量进行归一化,只有在可以推断绝对汗液量和浓度数据的情况下,才比PQN有明显的优势。为了能够做到这一点,我们需要知道一些关于动力学的信息以及我们感兴趣的代谢物的起始浓度。例如,当我们在之前的出版物中建模咖啡因网络时[20.,我们知道滞后所有代谢物的参数均为0,表明摄入的咖啡因总量(对应于\ (c_0 \))为200毫克。此外,我们知道咖啡因及其代谢物不是由人类合成的,并在我们的玩具模型中实施了相同的策略(对应于d).由于玩具模型被设计成类似于这样的新陈代谢,我们将这些信息转化为当前的研究。因此,我们假设玩具模型中的前4种代谢物是已知的\ (c_0 \),滞后,d参数。对应的\ (k_a \)和\ (k_e \)所有其他代谢物的参数设置为相同的界限\ ((0, 0, 0, 0) ^ {T} \ \文本勒(k_a、k_e c_0,滞后,d) ^ {T} \ \文本勒(3)3、3、5、15日^ {T} \ \文本)用于动力学数据生成。无花果。2显示浓度时间序列的例子,可以用修改的贝特曼函数和参数在拟合范围内描述。
真实手指汗液代谢组数据
真实世界的手指汗液数据是从参考文献中的研究C的37个时间序列测量中提取的。20.]。下载自MetaboLights (MTBLS2772和MTBLS2776)。
预处理。代谢组数据集分为两部分:靶向和非靶向。目标数据(即咖啡因、副黄嘌呤、可可碱和茶碱的大量时间序列数据)直接采用[36]。此数据可在GitHub (https://github.com/Gotsmy/finger_sweat).
对于非靶向代谢组学部分,使用ProteoWizard的msConvert工具(版本3.0.1928 -a2fc6eda4)将原始数据转换为mzML格式[37]。随后,使用MS-DIAL(4.70版)对样品中的代谢物和化合物进行非靶向检测[38]。首先对大多数(超过90%)样品中存在的几种化合物进行人工保留时间校正。这些化合物是单峰,在较早或较晚的保留时间内不存在异构体化合物(米/z697.755分5时57分;米/z564.359, 5.10分;米/z520.330, 4.85 min;米/z476.307, 4.58分;米/z415.253在4.28分钟;米/z371.227, 3.95分;米/z327.201, 3.56分钟;米/z3.13 min 283.175;米/z239.149分3.63分;米/z1.69分,166.080分;米/z159.113至1.19)。在此之后,进行无目标峰检测和自动对齐(在手动对齐之后),设置如下:质量精度MS1公差:0.005 Da,质量精度MS2公差:0.025 Da,保留时间开始:0.5 min,保留时间结束:6 min,执行保留时间校正:yes,最小峰高:1E5,质量片宽度:0.01 Da,平滑方法:线性加权移动平均,平滑级别:3次扫描,最小峰宽:5次扫描,对齐参考文件:C_D1_I_o_pos_ms1_1.mzML,保留时间公差:0.3 min, MS1公差:0.015 Da,空白去除系数:5倍变化)。没有进行空白减法,因为在每个样品中加入了内标准咖啡因,包括空白。峰丰度和元信息随对齐结果导出功能。
随后,我们排除了a中的异构体m / z差值小于({0.001} \ \ \ hbox {Da} \)且保持时间差小于({0.5} \ \ \ hbox{分钟}\).为了进一步减少潜在的背景特征,之后的保留时间特征({5.5} \ \ \ hbox{分钟}\)以及最小样本丰度的特征\(< 5\times \text {maximum blank abundance}\)(除了内标,咖啡因- d9)被排除在数据集中。这是以时间序列为基础的。因此,考虑正常化的非靶向代谢物的数量与平均值不同下午\ 343 \ 152 \)对于感兴趣的37个时间序列。
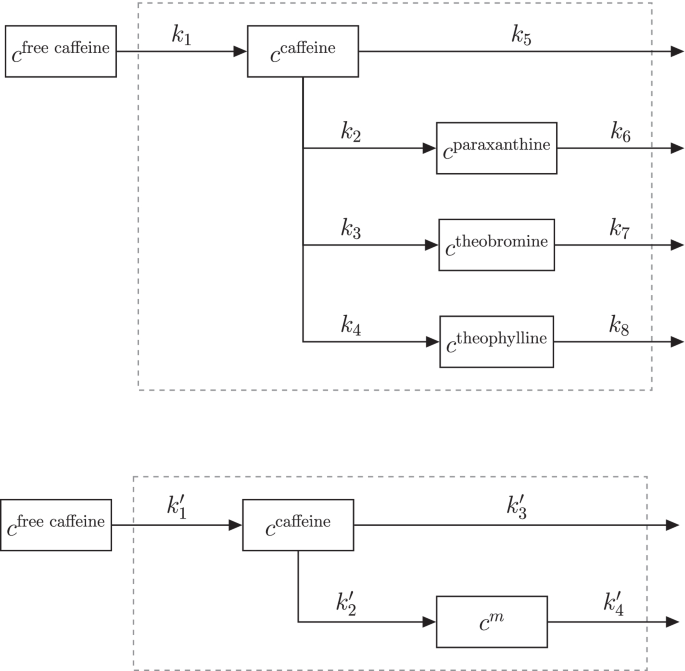
咖啡因吸收、转化为副黄嘌呤、可可碱和茶碱及其消除的完整网络(上图)和子网络(下图)。系统边界(虚线)代表人体。\(m \in \{\text{副黄嘌呤、可可碱和茶碱\}\)
尺寸效应归一化。在这个手指汗液数据集中,列出了目标代谢组学和非目标代谢组学的时间序列。四种目标代谢物(咖啡因、副黄嘌呤、可可碱和茶碱)的动力学是已知的。代谢物的反应网络如图顶部所示。4.简而言之,咖啡因首先被吸收,然后转化为三种降解代谢物。此外,所有四种代谢物都被排出体外。所有动力学都可以用一级质量作用动力学来描述[39,40]。
为了评估汗量归一化方法的性能,整个网络被分成三个子网络,每个子网络都含有咖啡因和一种降解代谢物(图2)。4底部面板)。在附加文件中给出了描述这种网络的一阶微分方程的解1:方程式。S2a和S2b。此外,下午\ 343 \ 152 \)非靶向代谢物时间序列被随机分成三个(几乎)大小相等的批次,每个批次被分配到一个子网。所有三个网络随后分别用PKM归一化\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\)方法的动力学参数调整到特定的反应网络(图2)。4底部面板)。随后,动力学常数(\ (k_1 \),\ (k_2 \),\ (k_3 \),\ (k_4 \))对37个测量浓度时间序列进行了估计。与原始出版物相比,拟合界限没有改变[20.]。
由于所有三个子网数据集都来自相同的手指汗液测量,因此潜在的动力学常数应该完全相同。吸收的动力学常数(\(k_a^\text {caf}= k_1'\))和消除(\(k_e^\text {caf}= k_2' + k_3'\)),我们使用它们的标准差来测试所测试的归一化方法的稳健性。
真实血浆代谢组数据
在Panitchpakdi等人的研究中[41摄取苯海拉明(DPH)后,在不同体液中测量代谢组的质量时间序列。在这里,我们关注血浆中测量的数据,其中包括DPH的丰度(已知动力学,校准曲线,药理学常数)及其三种代谢产物(已知动力学)和13526种未知动力学的非靶向代谢物的丰度。
预处理。峰面积数据从GNPS平台下载[42]。为了减少数据集中潜在的背景和/或噪声代谢物的数量,具有最小样本丰度的特征\(< 5\times \text {maximum blank abundance}\)在时间序列的基础上被排除在数据集中。因此,考虑正常化的非靶向代谢物的数量与平均值不同下午\ 1017 \ 114 \)对于我们感兴趣的10个时间序列。
尺寸效应归一化。我们假设四种代谢物(DPH、n -去甲基-DPH、DPH n -葡萄糖醛酸盐和DPH n -葡萄糖)的动力学可以用修饰的Bateman (Eq。10).代谢物的反应网络显示在附加文件中1:图S4。简而言之,DPH首先被吸收,然后通过未知的中间物转化为三种降解代谢物,这些代谢物依次被下游代谢或消除。\ (c_0 \)DPH的药理学常数为生物利用度、分布体积和DPH的剂量,根据原始出版物[41]。
类似于对手指汗液数据进行归一化,四种代谢物的完整网络被分成三个子网,其中只有一个共享的目标代谢物(DPH本身),另一个具有已知动力学的非目标代谢物(n -去甲基-DPH, DPH n -葡萄糖醛酸盐或DPH n -葡萄糖)1:图5)和三分之一下午\ 1017 \ 114 \)动力学未知的非靶向代谢物。为了保证模型在拟合过程中有更好的收敛性\ (\ widetilde {\ mathbf {M}} \)数据首先通过除以其代谢物的最大值缩放到0到1之间的值。这个因子可以再乘以\ (c_0 \)在标准化完成之后。此后,11\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\)模型被安装到比例模型上\ (\ widetilde {\ mathbf {M}} \)数据(\(\ well = 2\))。选择参数的范围,以便先前报告的估计[41]都在范围之内:\(0 \le k \le {5}\,\hbox {h}^{-1}\)为\ (\ {k_1, k_3 \} \),\(0 \le k \le {1}\,\hbox {h}^{-1}\)为\ (\ {k_2, k_4 \} \),{衰变时}\ \ (c_0 ^ \文本)根据原始出版物中报告的最大因子归一化,\(0\le c_0^m\le 300\)为\(m \in \{\)n -去甲基DPH, DPH n -葡糖苷,DPH n -葡萄糖\ \} \ ()和\(lag = d = 0\)以及\(0.01 \le V\le {0.03}\,\hbox {mL}\).
由于所有三个子网络数据集都来自相同的等离子体时间序列测量,因此DPH的潜在动力学常数应该完全相同。吸收的动力学常数(\(k_a^\text {DPH}= k_1'\))和消除(\(k_e^\text {DPH}= k_2'\))在所有三个子网中估计DPH,我们使用它们的标准差来测试PKM的鲁棒性\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\).
数据分析
归一化的优点。计算了两个拟合优度来分析测试方法的性能。RMSE为采样的汗液体积时间序列向量(\(\mathbf {V}^\text {true}\))减去拟合的汗液体积向量(\(\mathbf {V}^\text {fit}\)), rRMSE为抽样与拟合之比的标准差\ (\ mathbf {V} \)由均值归一化的向量。直观地说,RMSE是衡量拟合值与真实值之间的绝对差异有多大,另一方面,RMSE给出了拟合汗量相对于彼此的估计程度。RMSE和rRMSE的可视化描述显示在附加文件中1:图S6及其精确定义见3.3中的方程。
统计分析。通过计算检验拟合优度均值的显著性差异p值用非参数成对Wilcoxon符号秩检验[43) (SciPystats.wilcoxon函数(32])。显著性水平用*、**和***表示\(p \le 0.05\)分别为0.01和0.001。
结果

相对和绝对归一化性能。在最上面一行预测\(文本\{日志}_ {10}(C_ {j} (t_i; \ varvec {\ uptheta}) / C_ {j} (0; \ varvec {\ uptheta})) \)(\(i \in \{1,…,n_{\text {time points}}\}\),\(j \in \{1,…,{n_{\text {metabolites}}}\}\))绘制为真实的,潜在的\(文本\{日志}_ {10}(C_ {j} (t_i) / C_ {j} (0)) \).最下面一行显示了预测结果\ \ (V)作为真实的,潜在的函数\ \ (V).列表示不同的归一化模型(PQN, PKM\(_{\文本{最小}}\),以及MIX\(_{\文本{最小}}\)从左到右)。不是绝对的\ \ (V)可以从PQN计算,左下角的图省略。为了说明不同RMSE和rRMSE大小的影响(两者都是从\ \ (V)),我们显示了它们在100次重复中的平均值与R的比较\ (^ 2 \)从绘制的点计算出的值。直观上,rRMSE是对顶部行良好相关性的度量,而RMSE是对底部行(高R)良好相关性的度量\ (^ 2 \),低rRMSE/RMSE分别)
PKM与MIX的比较
综合数据模拟
为了测试不同归一化模型的性能,我们使用三种不同的方法(模拟v1, v2, v3)和五种不同的方法生成了100个合成数据集\ ({n_{\文本{代谢物}}}\)(4、10、20、40、60)各,其中底层\ (\ mathbf C {} \),\ \ (V),\ (\ varvec {\ upepsilon} \)价值是已知的。模拟v1, v2和v3的不同之处在于\ (\ mathbf C {} \)生成(动态、随机、分别从真实数据集采样)。为了量化归一化模型的性能,我们使用了两个衡量归一化优度的指标来分析结果:RMSE和rRMSE。
为了可视化获得的归一化性能,我们绘制了模拟v3和\({n_{\文本{代谢物}}}= 60\)在无花果。5为三种归一化模型(从左至右列,PQN, PKM\(_{\文本{最小}}\),以及MIX\(_{\文本{最小}}\)).最上面一行显示了预测结果\(文本\{日志}_ {10}(C_ {j} (t_i; \ varvec {\ uptheta}) / C_ {j} (0; \ varvec {\ uptheta})) \)(即每种代谢物的浓度j在每个时间点我除以时间0时的浓度作为真实值的函数\(文本\{日志}_ {10}(C_ {j} (t_i) / C_ {j} (0)) \)值。它说明了一种代谢物在所有时间点的相对丰度的相关性。良好的相关性(即高R)\ (^ 2 \)),如PQN和MIX所示\(_{\文本{最小}}\)导致rRMSE测量值较低。在图的下一行。5预测值的绝对值\ \ (V)是真实的函数吗\ \ (V).很明显,良好的绝对值相关性导致RMSE测量值较低。
在下面的部分中,我们将分别关注RMSE和rRMSE的大小,因为它们都是根据预测计算的\ \ (V)直接。注意,对于PQN,没有绝对\ \ (V),因此不计算RMSE。
代谢物数量的影响。我们跟踪了不同代谢物数量归一化方法的均方根误差和均方根误差(\ ({n_{\文本{代谢物}}}\))来研究这些方法在不同数量的可用信息下的表现。的函数,概述了它们的归一化度量的良度\ ({n_{\文本{代谢物}}}\)图中给出了采样的动态数据(图A、B)、完全随机数据(图C、D)和采样的真实数据子集(图E、F)。6.

合成数据模拟的归一化度量的优度。给出了不同汗液量归一化模型的RMSE(左列)和rRMSE(右列)的100个重复的平均值。模拟v1、v2和v3的结果分别显示在第一行、第二行和第三行中。误差条表示重复的标准差。对于PQN方法,无法计算RMSE
PKM\(_{\文本{全}}\)它符合所有可能代谢产物的动力学功能(\(\ well = {n_{\text{代谢物}}}\))表现良好(低RMSE,低rRMSE)\ (\ mathbf C {} \)数据来源于一个动力学函数(仿真v1,图1)。6A, B)。然而,当底层数据不是来源于动态时间序列时(模拟v2,图2)。6C, D,它的性能大大降低。为11\(_{\文本{全}}\)这与均方根误差(RMSE)的增加相似\(0.19 \pm {0.08}\,\upmu \hbox {L}\)来\(0.64 \pm {0.16}\,\upmu \hbox {L}\)为\({n_{\文本{代谢物}}}= 60\))以及rRMSE(来自\(0.08 \pm 0.02\)来\(0.28 \pm 0.14\)为\({n_{\文本{代谢物}}}= 60\)).
另一个观察结果是PQN的行为。随着rRMSE的增加,rRMSE接近于0\ ({n_{\文本{代谢物}}}\),而不关心底层数据是如何生成的。
有趣的是,模拟v3的结果位于模拟v1和v2的结果之间。在比较PKM的性能时,这一点尤为明显\(_{\文本{全}}\)在无花果。6.这样的结果表明,并非所有测量的非靶向代谢物都是完全随机的,但有些可以用改良的贝特曼函数来描述。这导致假设,在汗液量归一化之后,真实的手指汗液数据(从中采样v3值)具有发现未知动力学的高潜力。
所有归一化方法的RMSE和rRMSE的确切数字\ ({n_{\文本{代谢物}}}\)在附加文件中给出1:表S3、表S4。此外,将归一化方法的RMSE和rRMSE与PKM结果进行两两比较\(_{\文本{最小}}\)绘制在附加文件1图S7。

模拟v3的RMSE度量\({n_{\文本{代谢物}}}= 60\).采用双侧Wilcoxon符号秩检验,在100个配对重复上计算方法间的显著性
统计测试。在\({n_{\文本{代谢物}}}= 60\)归一化措施的优度开始趋于平缓,我们进一步调查了这种情况的统计显著性。我们使用双侧非参数Wilcoxon符号秩检验来比较被检验模型之间RMSE和rRMSE的两两差异。p-所有组合的值在附加文件中给出1表S5和表S6。
无花果。6已经指出,在RMSE和rRMSE中,MIX的总体性能最好\(_{\文本{最小}}\)模型。为\({n_{\文本{代谢物}}}= 60\)它显著优于其他所有方法的RMSE(图2)。7).此外,混合\(_{\文本{最小}}\)在rRMSE中的性能至少等于或优于所有其他测试方法(附加文件1表S6),但有一个例外:MIX的rRMSE比较\(_{\文本{最小}}\)和PQN在模拟v1中有显著差异(\(p = 0.0029\)),但是,rRMSE的绝对值仍然非常相似(\(0.049下午\ 0.010 \)和\(0.047下午\ 0.009 \)分别)。与以前使用的PKM相比\(_{\文本{最小}}\)[20.],即MIX的均方根误差\(_{\文本{最小}}\)提高了\(73 \pm {10}\,\%\), rRMSE由\(43 \pm {12}\,\%\)(附加文件1:图S7)。与图1类似。7对于仿真v3,仿真v1和仿真v2的结果显示在附加文件中1:无花果。分别是S8和S9。
使用双侧版本的Wilcoxon符号秩检验来检验多种归一化方法之间的任何差异。很明显,MIX\(_{\文本{最小}}\)我们使用单侧版本的Wilcoxon有符号秩检验来验证混合是否显著降低RMSE和rRMSE\(_{\文本{最小}}\)与其他归一化方法相比。由此产生的p-value列在附加文件中1表S7。再一次,混合\(_{\文本{最小}}\)在任何模拟中,除PQN外,在RMSE和rRMSE中都明显优于所有其他测试方法。
因此,我们得出结论,通过MIX使汗液量正常化\(_{\文本{最小}}\)方法减小了估计值的误差\ \ (V)与其他测试方法相比。与PKM相比,MIX\(_{\文本{最小}}\)的优点是,如果代谢物的浓度时间序列可以用修改的Bateman函数来描述(即模拟v1, v2 v3对其性能影响很小),则其性能不会发生变化。因此,如果不能保证此属性,则特别有利。
计算性能
代谢组学数据集的分析通常是一个计算详尽的过程。在(预)处理中有几个步骤需要执行,其中许多需要持续几个小时。因此,计算时间可以迅速叠加到很大的数字。规范化模型也不例外。作为\ ({n_{\文本{代谢物}}}\)在药代动力学模型增加的情况下,优化药代动力学模型的时间可能会受到限制。因此,我们研究了不同方法和不同代谢物数量的一次时间序列归一化的平均时间。

仿真v3中优化一个规范化模型的时间(以秒为单位)。误差条表示100次重复之间归一化次数的标准差
一个优化步骤所花费的计算时间是的函数\ ({n_{\文本{代谢物}}}\)如图所示。8对于仿真v3。对于某些归一化模型,它增加了,但不是对所有模型都增加了,也不是相等的。在所研究的范围内,PQN每次归一化保持在1秒以下,而PKM\(_{\文本{全}}\)归一化时间从\(1.6 \pm {1.1}\,\hbox {s}\)对于一个有4种代谢物的模型\(110 \pm {44}\,\hbox {s}\)60种代谢物。MIX的归一化时间也类似\(_{\文本{全}}\)最大限度地\(19 \pm {22}\,\hbox {s}\)为\({n_{\文本{代谢物}}}= 60\).与完整模型所需的计算能力呈指数级增长形成鲜明对比的是最小模型。他们优化的时间几乎保持不变(\(< {3}\,\hbox {s}\))在所研究的代谢物范围内(附加文件)1表8)。
这里我们演示一下MIX\(_{\文本{最小}}\)不仅在归一化性能上优于其他已测试模型,而且在计算可行性上也优于其他模型。我们假设,即使包含数千种非靶向代谢物的数据集也会对其速度产生轻微影响。
PQN与MIX的比较

噪声数据占比对PQN计算误差的影响。图A显示了方差系数的变化\(\mathbf {Q}^\text {PQN}\)(y轴)为噪声分数(\ (fn \)(y轴与颜色条具有相同的刻度标签)增加。图B显示了计算的误差大小文本\(问^ \ {PQN} \)为真\ \ (V)平均缩放(x轴)和标准缩放(y轴)。点的颜色与颜色条中描述的噪声分数有关
噪声对PQN的影响
在非靶向代谢组学中,通常很难区分来自实际基质的代谢物或来自污染的代谢物。由于PQN在计算中包含了所有非目标代谢物,由于污染产生的代谢物的折叠变化与汗液量无关,从而改变了商的底层分布,因此可能会成为一个问题。因此,我们研究了来自污染(即噪声数据)的代谢物的不同组分的影响。此外,我们还测试了\(\mathbf {Q}^\text {PQN}\)数值可以抵消噪声带来的误差。
数字9A演示了在有噪声的原始数据上使用概率商归一化的问题。当存在噪声时,尺寸效应的方向仍然可以解释,但尺寸效应的绝对值减小。因此,在图1中。9的变异系数(即标准差除以均值)\({\textbf {Q}}^\text {PQN}\)是对一个综合生成的时间序列的估计规模效应的平均值的度量。作为噪声(\ (fn \)(x轴)增大时,变异系数急剧减小,趋于0\(f_n \rightarrow 1\).
数字9B显示了标度方法抵消上述变异系数降低的性能。平均标度误差(x轴)和标准标度误差(y轴)由式计算。17互相密谋。当\(f_n \le 0.05\),平均缩放优于标准缩放。不过,此后标准扩大了文本\(问^ \ {PQN} \)比平均缩放版本误差更小。
当合并\(\mathbf {Q}^\text {PQN}\)值,重要的是要纠正由噪声引入的误差。结果表明,标准标度减小了噪声对计算的不利影响\(\mathbf {Q}^\text {PQN}\),我们在整个MIX归一化研究中使用标准缩放。此外,这一结果强调了标准缩放在生物数据集中的良好性能[44]。

PQN与MIX的rRMSE比较\(_{\文本{最小}}\)对具有不同噪声分数的数据。PQN与MIX的rRMSE差异有统计学意义\(_{\文本{最小}}\)单侧两两Wilcoxon sign -rank检验
含噪声的合成数据模拟
用于断面分析的综合数据PKM与MIX的比较不含任何被归类为噪音的代谢物,即他们的\ (\ widetilde {\ mathbf {M}} \)不受规模效应的影响(式。15).然而,这并不一定是一个现实的假设,因为在代谢组测量中有许多污染物来源。嘈杂的代谢物可以通过生物手段引入(例如,在汗液测量中,代谢物不是来自汗液,而是来自皮肤表面)[45]或实验性处理[46]。如图所示。9,数据中的噪声会对PQN的性能产生负面影响。节结果中PQN的良度PKM与MIX的比较可能被高估了。
为了更准确地了解PQN和MIX归一化的优点\(_{\文本{最小}}\),我们在合成数据上测试了它们的性能,\ (fn \).为了做到这一点,我们创建了100个从真实数据(即模拟v3)中采样的合成数据的复制,用于10个等距噪声分数,范围从\(n = 0\)来\(f_n = 0.9\)与\({n_{\文本{代谢物}}}= 60\).在所有模拟数据中,只有非靶向代谢物受到噪声引入的影响,因为我们假设对于靶向代谢物(即,\(\ well = 4\))有已知的药代动力学行为,人们可以高度确信测量结果不是来自污染物。
PQN和MIX的rRMSE\(_{\文本{最小}}\)如图所示。10.仅当合成数据集MIX中存在零噪声时\(_{\文本{最小}}\)并没有改善PQN。但是,随着噪声分数的增加,MIX\(_{\文本{最小}}\)在rRMSE方面明显优于PQN。的p-所有噪声分数的值在附加文件中列出1表S9。
PQN与MIX的rRMSE差异\(_{\文本{最小}}\)在无花果。10与图中平均尺度误差与标准尺度误差之差有关。9B. PQN不能单独利用标准缩放的改进性能\(\text {Std}(T(\mathbf {V}))\)必须以其计算而闻名(式2)。17 b).但是,当使用MIX进行规范化时\(_{\文本{最小}}\),\(\text {Std}(T(\mathbf {V}))\)可以从模型的药代动力学部分估计(Eq。9 c),显著提高了产品质量。

用brunmaair等人,2021年的手指汗液(左柱)和血浆(右柱)数据验证方法[20.]和Panitchpakdi等人,2021 [41分别)。在A至D组,咖啡因和苯海拉明的吸收和消除常数的标准偏差({caf} \ \ (k_a ^ \文本),{caf} \ \ (k_e ^ \文本),{衰变时}\ \ (k_a ^ \文本),{衰变时}\ \ (k_e ^ \文本)),绘制了三个模型子网之间的关系。每种方法的点数对应于两个数据集中存在的浓度时间序列的数目(即,汗液和血浆分别为37和10)。采用单侧Wilcoxon符号秩检验检验显著性差异。图E和图F显示了从三个不同的子网络绘制的咖啡因和DPH的估计浓度时间序列。这些系以子网络中存在已知动力学的第二代谢物命名;然而,他们都指的是C咖啡因和DPH曲线的颜色和曲线之间的面积表示用PKM归一化的结果\(_{\文本{最小}}\)或混合\(_{\文本{最小}}\)分别
应用于实际数据
咖啡因网络
在此之前,我们在摄入单剂量咖啡因后确定并量化了四种代谢物(咖啡因、副黄嘌呤、可可碱和茶碱)的时间序列[20.]。为了研究归一化模型在真实手指汗液数据集上的性能,我们对所有测量数据进行了拆分\ (\ widetilde {\ mathbf {M}} \)时间序列分成三个部分,每个部分包含对目标代谢物,只有一个是所有人共享的,即咖啡因(比较图2)。4顶部和底部网络)。随后我们安装了PKM\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\)模型(\(\ well = 2\))和适应动力学(方法部分)真实手指汗液代谢组数据)通过三个子数据集。由于代谢物子网络的性质(图2)。4下图)可以计算出描述咖啡因吸收和消除的两个动力学常数(\(k_a^\text {caf}= k_1'\)和\(k_e^\text {caf}= k_2' + k_3'\))。由于所有三个子网的数据都是在同一个实验中测量的,我们可以假设这些常数的基本真理是相同的。因此,通过比较动力学常数的标准差,可以推断出归一化方法的性能。
在图A和B面板。11,一次测量的拟合动力学常数的标准差\ (\ widetilde {\ mathbf {M}} \)时间序列图。图A显示了咖啡因吸收常数的标准差,{caf} \ \ (k_a ^ \文本), PKM的\(_{\文本{最小}}\)明显大于MIX\(_{\文本{最小}}\)模型(\(p = 5.8\乘以10^{-4},n = 37\)单侧Wilcoxon sign -rank检验)。同样,MIX的标准差大小也显著减小\(_{\文本{最小}}\)与之前发表的PKM\(_{\文本{最小}}\)模型(\(p = 1.5~10^{-5}\))来持续消除咖啡因,{caf} \ \ (k_e ^ \文本)(面板B,图2)11).
在图E面板中。11,其中一个例子是规范化的C汗水中咖啡因的时间序列被描述为适合所有三个具有PKM的子网\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\),分别。所选的时间序列说明了PKM之间标准差差异的中位数\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\)从图A和B面板。11.被包围的区域CMIX的s\(_{\文本{最小}}\)模型比PKM小\(_{\文本{最小}}\).
我们强调,在我们最初的研究中,咖啡因降解直接产生副黄嘌呤、可可碱和茶碱;因此,药代动力学参数\(k_2, k_3, k_4\)明确地联系在一起[20.]。因此,动力学网络类似于该代谢途径的特定动力学(图2)。4前面板)。相反,在前面的章节中,我们假设潜在的通路结构是未知的。因此,参数没有链接,这意味着参数的约束较少。然而,在本节中,我们证明了从PKM切换到MIX模型所发现的根本改进也可以再次转化为更具体的代谢网络(图2)。4底部面板)。为了支持这一论点,我们展示了MIX的适用性\(_{\文本{最小}}\)真实手指汗液数据集的归一化方法。实际数据的结果强调了在合成数据集上模拟的有效性。他们表明,特别是当已知的代谢网络很小时,MIX\(_{\文本{最小}}\)模型显著提高了归一化的鲁棒性,从而从手指汗液时间序列测量中推断出动力学常数。
苯海拉明网络
在最初的研究中[41],作者测量了单剂量苯海拉明(DPH)应用后血浆中的时间序列丰度。\ (\ widetilde {\ mathbf {M}} \)从靶向DPH(已知药理学常数,已知动力学)以及非靶向代谢产物(n -去甲基-DPH, DPH n -葡萄糖醛酸盐,DPH n -葡萄糖,已知动力学)和其他几种非靶向代谢物(未知动力学)报道。与汗液类似,虽然不那么明显,血浆也受到由生物手段或分析前样品处理引入的尺寸效应(即测量中的系统误差)的影响[47,48]。因此,我们使用报告的数据作为验证MIX性能的第二个真实数据集\(_{\文本{最小}}\).验证的执行类似于咖啡因的研究,其中一个完整的网络(附加文件)1:图S4)被分成三个子网(附加文件1:图S5,详见“方法”章节。真实血浆代谢组数据”)。
在图的C和D面板中。11,一次测量的拟合动力学常数的标准差\ (\ widetilde {\ mathbf {M}} \)并举例说明了三个拟合的子网。的标准差{衰变时}\ \ (k_a ^ \文本)的11\(_{\文本{最小}}\)明显大于MIX\(_{\文本{最小}}\)(\(p = 2,0\乘以10^{-3},n=10\),单侧Wilcoxon sign -rank检验,图C)。标准差也有类似的显著下降{衰变时}\ \ (k_e ^ \文本)(\(p = 3.2~10^{-2}\)选D。
在图F面板中。11,其中一个例子是规范化的C等离子体中DPH的时间序列被描述为适合所有三个具有PKM的子网\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\),分别。选择时间序列是因为它最接近PKM之间标准差差异的中位数\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\).可见,被包围的区域C混合的结果\(_{\文本{最小}}\)模型比PKM模型小\(_{\文本{最小}}\).
该验证说明了本研究中提出的归一化模型在独立于所述方法开发的数据集上的性能。血浆验证研究的结果与手指汗液研究的结果相似;再一次,混合\(_{\文本{最小}}\)提高尺寸效应归一化的稳健性(即减少标准差)。
尽管标准差显著降低{衰变时}\ \ (k_e ^ \文本)与混合\(_{\文本{最小}}\)与PKM相比\(_{\文本{最小}}\)、混合\(_{\文本{最小}}\)也产生了一个异常值(图2)。11D).出现这种异常值的原因是,在极少数情况下,MIX\(_{\文本{最小}}\)由于收敛问题,无法检测到任何大小影响(其他文件?1:图S10A)。为了研究这些结果,我们执行了合成数据模拟(附加文件)1:图S10B)。在那里,我们发现MIX的这种行为\(_{\文本{最小}}\)能不能观察到两者何时不同\ (\ mathbf {V} \)向量被应用于\ \)(\拼和\ \ l形+ \)代谢物。因此,我们假设MIX明显可见的功能障碍\(_{\文本{最小}}\)检测大小效应(即估计的方差)\ (\ mathbf {V} \)接近于0)向科学家表明,在这样的数据集中,大小效应可能不是一个主要问题。例如,在这个特定的血浆时间序列测量中,与其他误差源相比,尺寸效应可能太小,无法由MIX识别\(_{\文本{最小}}\).
总之,通过这一验证,我们表明,只要修正的Bateman函数能够很好地描述所测量的动力学,并且尺寸效应足够大,可以检测到,本研究中实现的广义归一化模型就可以直接用于实际数据的归一化。
讨论
在本研究中,我们提出了一个广义的PKM归一化模型框架,该框架在文献[20.]。此外,我们扩展了现有的模型,纳入了非靶向代谢物信息,称为MIX模型。这两个模型都是用Python实现的,可以在GitHub上获得https://github.com/Gotsmy/sweat_normalization.
在合成数据集上测试了归一化方法的质量。合成数据集是必要的,因为如果不从根本上改变上述(手指)汗液采样方法,就不可能获得验证数据[20.]。然而,我们采用了三种不同的合成数据生成方法(v1、v2、v3)来确保合成数据集尽可能接近真实数据。我们发现当\({n_{\文本{代谢物}}}\ge 60\)、混合\(_{\文本{最小}}\)性能与所有其他经过测试的规范化方法一样好或更好。
尽管真正的\ \ (V)值仍然未知,真实手指汗液数据可以作为标准化方法相对稳健性的验证。在那里,混合\(_{\文本{最小}}\)显著优于PKM\(_{\文本{最小}}\).MIX估算的动力学常数方差减小\(_{\文本{最小}}\)可能源于这样一个事实文本\(问^ \ {PQN} \)只要足够多,三个子集差别不大\({n_{\文本{代谢物}}}= 60\)存在于每个子集中。另一方面,由于只有很少的数据点用于PKM\(_{\文本{最小}}\)优化后,两个目标代谢物测量质量中的一个的小误差有很大的可能改变归一化结果。
此外,PKM的性能\(_{\文本{最小}}\)和混合\(_{\文本{最小}}\)对血浆数据集进行比较,该数据集取自一项独立于任何用于标准化模型开发的测量的研究。在那里,我们能够演示来自PKM的相同改进\(_{\文本{最小}}\)混合\(_{\文本{最小}}\)在归一化鲁棒性。此外,我们表明,在本研究中,作为Python类实现的广义归一化模型可以很容易地用于尺寸效应归一化,几乎不需要额外的编码。
概括地说,拟议的MIX\(_{\文本{最小}}\)模型与其他测试方法相比有几个关键的优点。
混合\(_{\文本{最小}}\)显著优于PKM\(_{\文本{最小}}\)相对(rRMSE);\(-43 \pm {12}\,\%\))和绝对值(RMSE,\(-73 \pm {10}\,\%\))错误,只有60种非靶向代谢物被用作附加信息(图2)。7).
混合\(_{\文本{最小}}\)对于非靶向代谢物是否遵循易于描述的动力学浓度曲线是不变的(图2)。6).
无噪声,MIX\(_{\文本{最小}}\)在相对丰度上的表现与PQN一样好,但另外,它估计的绝对值\ \ (V),类似于药代动力学(PKM)模型(图2)。6).
当噪声存在时混合\(_{\文本{最小}}\)相对丰度也优于PQN(图2)。10).
混合\(_{\文本{最小}}\)在这一原理论证研究中表现良好;此外,它可以作为进一步改进的基础。首先,不同的、更复杂的统计归一化方法(例如,EigenMS [27])可以作为模型的PQN部分的输入。其次,描述不同代谢物的不确定性的贝叶斯先验可以实现\λ(\ \)参数以与参考文献[中讨论的类似的方式49]。
引人注目的是,结果表明,对于所有测试的归一化方法,一旦原始信息中存在60种代谢物,RMSE和rRMSE值就会变平。这表明所提出的归一化模型,特别是MIX\(_{\文本{最小}}\),甚至可以应用于生物基质或分析方法,只测量60种化合物。
虽然混合\(_{\文本{最小}}\)特别是考虑到汗液量标准化,它可以很容易地适用于其他生物基质,例如血浆(图2)。11).
结论
在本研究中,我们描述并定义了MIX代谢组学时间序列归一化模型,并将其与PKM进行了比较。随后,我们阐述了MIX的几个优点\(_{\文本{最小}}\)模型和先前发布的归一化方法。我们相信,这将进一步提高对手指汗液和其他传统和非传统生物液体进行代谢组学研究的可靠性。然而,我们承认需要进行更彻底的调查,使用几种更量化的代谢物和确定的汗液量的数据集来评估所提出方法的全部潜力。
数据和材料的可用性
所有分析(除非另有说明)都在Python 3.7中执行,严重依赖于NumPy [50],熊猫[51]和SciPy [32]。在GitHub上可以找到用于模拟的代码、创建图形的脚本以及原始和生成的数据https://github.com/Gotsmy/sweat_normalization在GNU GPL版本3许可下。
缩写
- 文本\(现代\{样本}\):
-
取样皮肤面积
- b:
-
部分修改贝特曼函数
- \(C, \mathbf {C}\):
-
潜在浓度(矢量)
- \ (c_0 \):
-
动力学参数
- d:
-
动力学参数
- F:
-
修改贝特曼函数
- \ (fn \):
-
噪音分数
- 我:
-
时间点指数
- j:
-
代谢物指数
- k:
-
动力学参数
- \ \)(\拼:
-
用于动力学拟合的代谢物
- \ \ l形+ \):
-
不用于动力学拟合的代谢物
- \ (\ mathcal {L} \):
-
损失
- l:
-
损失函数
- 滞后:
-
动力学参数
- \(\ widdetilde {M}, \ widdetilde {\mathbf {M}}\):
-
测量质量(矢量)
- {ref} \ \ (M ^ \文本):
-
PQN的参考质量
- 米/z:
-
质量电荷比
- \ ({n_{\文本{代谢物}}}\):
-
代谢物数量
- \(n_{\text{时间点}}\):
-
时间点数
- p:
-
p价值
- 文本\ (q_ \{汗}\):
-
出汗率
- \(问^ \ mathbf C {} \):
-
两个样本的中位数浓度折叠变化
- \(问^ \ mathbf {M} \):
-
两个样本的中位数质量折叠变化
- 文本\(问^ \ {PQN} \),\(\mathbf {Q}^\text {PQN}\):
-
由PQN计算的归一化商(向量)
- R\ (^ 2 \):
-
决定系数
- 推定:
-
标准化优度的相对度量
- RMSE:
-
归一化优度的绝对度量
- Std:
-
标准偏差
- T:
-
转换函数
- t:
-
时间
- \ \ (V),\ (\ mathbf {V} \):
-
采集(汗液)体积(载体)
- \ (V ^文本{ref}} {\ \):
-
PQN参考卷
- Var:
-
方差
- V1 v2 v3:
-
合成数据集
- Z:
-
尺度函数
- \ (\ varvec {\ upepsilon} \):
-
实验误差矢量
- \ (\ varvec {\ uptheta} \):
-
拟合的动力学参数向量
- \λ(\ \):
-
损失加权参数
- \ \(τ\):
-
收集一个样本的时间
参考文献
张建军,张建军,张建军,张建军,张建军,张建军,张建军,张建军,张建军,张建军,张建军,张建军,张建军,张建军。科学通报,2020;10(1):1 - 7。
Delgado-Povedano M, Calderón-Santiago M, de Castro ML, prigo - capote F.适度运动后人体汗液代谢组学分析。Talanta。2018;177:47 - 65。
刘建军,刘建军,刘建军,刘建军,刘建军。基于代谢组学的生活方式监测方法研究进展[J]。中国生物医学工程学报,2013 (1):1 - 13
Czerwinska J, Jang M, Costa C, Parkin MC, George C, Kicman AT, Bailey MJ, Dargan PI, Abbate V.液相色谱-串联质谱和纸喷雾质谱法检测甲氧麻黄酮及其代谢物的对照研究。分析师。2020;145(8):3038 - 48。
Calderón-Santiago M, Priego-Capote F, Turck N, Robin X, Jurado-Gámez B, Sanchez JC, De Castro MDL。人体汗液代谢组学用于肺癌筛查。生物化学学报,2015;37(6):581 - 592。
崔霞,张丽,苏刚,Kijlstra A,杨鹏。眼部白塞病的汗液代谢特征分析。中华医学杂志,2011;17:1079 - 1082。
Harshman SW, Browder AB, Davidson CN, Pitsch RL, Strayer KE, Schaeublin NM, Phelps MS, O 'Connor ML, Mackowski NS, Barrett KN等。营养补充对汗液代谢组学含量的影响:一项概念验证研究。前沿化学。2021;9:255。
胡建军,陈建军,陈建军。汗液代谢组学分析中汗液采集标准化的挑战。临床生物化学学报,2017;38(1):13。
Harshman SW, Strayer KE, Davidson CN, Pitsch RL, Narayanan L, Scott AM, Schaeublin NM, Wiens TL, Phelps MS, O 'Connor ML等。汗液代谢组学生物标志物发现的速率正常化。天津大学学报。2021;223:121797。
Kuwayama K, Tsujikawa K, Miyaguchi H, Kanamori T, Iwata YT, Inoue H.咖啡摄入后指尖提取咖啡因及其代谢物的时间过程测量:指纹检测药物的初步研究。中国生物医学工程学报,2013;35(6):945 - 952。
Kuwayama K, Yamamuro T, Tsujikawa K, Miyaguchi H, Kanamori T, Iwata YT, Inoue H.药物给药后指尖转移药物和代谢物的时间过程测量:指纹对药物检测的有用性。法医毒物,2014;32(2):235-42。
汗腺功能生理学:出汗和汗液成分在人体健康中的作用。温度。2019;6(3):211 - 59。
李建军,陈斌,李建军,李建军,李建军,李建军,李建军,李建军。一种可穿戴式热调性汗液检测方法。生物医学工程学报,2011;12(1):1 - 13。
Taylor NA, Machado-Moreira CA。人体静息和运动时经皮失水、汗腺密度、汗液分泌率和电解质组成的区域差异。中华医学杂志,2013;2(1):4。
Ando H, Noguchi R.手掌出汗反应和中枢神经系统活动对全身振动频率的依赖。[J]劳动环境与卫生,2003:216-219。
钟波,姜凯,王磊,沈刚。可穿戴式失汗量测量装置:从失汗作用到先进机制与设计。科学通报,2012,21(2):357 - 357。
Harshman SW, Pitsch RL, Smith ZK, O 'Connor ML, Geier BA, Qualley AV, Schaeublin NM, Fischer MV, Eckerle JJ, Strang AJ,等。运动诱导汗液的蛋白质组学和代谢组学特征用于人体性能监测:一项试点研究。PLoS ONE。2018; 13(11): 0203133。
王志强,王志强,王志强,王志强,王志强,王志强,王志强,等。分泌汗腺的微流体,包括生物标志物的分配、运输和生物传感意义。生物微流体学报,2015;9(3):031301。
杜强,张勇,王军,常军,王安,任旭,刘波。指纹图谱中17种降糖药物的超高效液相色谱/串联杂交三重四极杆线性离子阱质谱定量分析。快速共质谱。2022;36(1):9199。
Brunmair J, Gotsmy M, Niederstaetter L, Neuditschko B, Bileck A, Slany A, Feuerstein ML, Langbauer C, Janker L, Zanghellini J,等。手指汗液分析可实现人体短间隔代谢生物监测。生物医学工程学报,2011;12(1):1 - 13。
Filzmoser P, Walczak B.在识别生物标志物的数据规范化步骤中可能出现什么问题?[J] .中国生物医学工程学报,2014;32(2):391 - 391。
马苏库。应用统计学研究中的抽样技术和样本量的确定:综述。国际经济管理,2014;2(11):1-22。
Choi J, bandokar AJ, Reeder JT, Ray TR, Turnquist A, Kim SB, Nyberg N, Hourlier-Fargette A, Model JB, Aranyosi AJ,等。柔软,皮肤集成多功能微流控系统,用于汗液生物标志物和温度的准确比色分析。传感器学报,2019;4(2):379-88。
金某人,古永锵J, Yoon J, Hourlier-Fargette,李B,陈年代,乔,崔J,哦,y,李G, et al。用于测量汗液中氨和乙醇浓度的集成酶分析的软皮肤界面微流控系统。实验室芯片,2020;20(1):84-92。
Ragan TJ, Bailey AP, Gould AP, Driscoll PC。体积测定与两个标准允许绝对定量和改进的化学计量分析代谢物的核磁共振从亚微升样品。化学工程学报,2013;35(2):1 - 4。
Warrack BM, Hnatyshyn S, Ott K-H, Reily MD, Sanders M, Zhang H, Drexler DM.尿液代谢组学分析归一化策略。中国生物医学工程学报,2009;31(5):557 - 557。
刘建军,刘建军,刘建军,刘建军。代谢组学数据用特征谱归一化。PLoS ONE。2014; 9(12): 116221。
李建军,李建军,李建军,等。基于概率商归一化的生物混合稀释分析方法。核磁共振代谢组学的应用。化学工程学报。2006;32(1):1 - 4。
李波,唐杰,杨青,崔霞,李松,陈生,曹青,薛伟,陈宁,朱峰。基于lc/ms的非靶向代谢组学分析中数据驱动归一化方法的性能评价及在线实现。科学通报,2016;6(1):1 - 13。
邓文杰,张建军,张建军,张建军,张建军,张建军,张建军。非目标uhplc-ms代谢组学数据处理方法:标准化,缺失值输入,转换和缩放的比较研究。代谢组学。2016;12(5):93。
马西多,马西帕拉南,马西帕拉南,马西帕纳,马西帕纳,马西纳,马西纳,马西纳,马西纳,马西纳,马西纳,马西纳,马西纳。中国化学学报,2017;3(8):904-13。
Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, burrovski E, Peterson P, wecesser W, Bright J,等。Scipy 1.0: python科学计算的基本算法。自然科学学报,2020;17(3):261-72。
加勒特。重新审视贝特曼函数:对定量表达的关键重新评估,以表征一室体模型中的浓度作为一阶侵入和一阶消除的时间函数。中国生物医学工程学报,2009;22(2):103-28。
刘建军,刘建军,刘建军,等。基于多聚类的非靶向lc-ms代谢组学数据校正方法。代谢组学。2016;12(11):1-13。
kvasnika A, Friedecký D, tich
Brunmair J, Gotsmy M, Niederstaetter L, Neuditschko B, Bileck A, Slany A, Feuerstein ML, Langbauer C, Janker L, Zanghellini J,等。手指汗液分析可实现人体短间隔代谢生物监测。https://doi.org/10.5281/zenodo.5222967.
Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J,等。质谱和蛋白质组学的跨平台工具包。生物工程学报,2012;30(10):918-20。
Tsugawa H, Ikeda K, Takahashi M, Satoh A, Mori Y, Uchino H, Okahashi N, Yamada Y, Tada I, Bonini P,等。脂质体图集在ms-dial 4。生物工程学报,2020;38(10):1159-63。
张晓明,张晓明,张晓明,等。麻黄碱、去甲麻黄碱和咖啡因在人体中的作用机制。中华临床医学杂志,2005;59(3):335-45。
Kamimori GH, Karyekar CS, Otterstetter R, Cox DS, Balkin TJ, Belenky GL, Eddington ND。在正常健康志愿者中,口香糖和胶囊对咖啡因的吸收率和相对生物利用度。中华临床医学杂志,2002;32(1):1 - 4。
Panitchpakdi M, Weldon KC, Jarmusch AK, Gentry EC, Choi A, Sepulveda Y, Aguirre S, Sun K, Momper JD, Dorrestein PC等。非侵入性皮肤取样检测人体系统给药。科学通报,17(7),e0271794。
Panitchpakdi M, Weldon KC, Jarmusch AK, Gentry EC, Choi A, Sepulveda Y, Aguirre S, Sun K, Momper JD, Dorrestein PC等。非侵入性皮肤取样检测人体系统给药。https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=deee382b163f4441afea5fda4b2a2bcf.2021年12月20日访问。
Wilcoxon F.排名法的个体比较。生物识别学报。1945;1(6):80-3。
van den Berg RA, Hoefsloot HC, Westerhuis JA, Smilde AK, van der Werf MJ。集中、缩放和转换:提高代谢组学数据的生物信息含量。生物医学工程学报,2006;7(1):1 - 15。
贝克LB,沃尔夫AS。决定汗液组成的生理机制。中国生物医学工程学报,2014;31(4):559 - 561。
da Silva RR, Vargas F, Ernst M, Nguyen NH, Bolleddu S, Del Rosario KK, Tsunoda SM, Dorrestein PC, Jarmusch AK。通过肯德里克质量滤波器计算去除具有重复单元的不希望的质谱特征。质谱学报,2018,30(2):268-77。
Kamlage B, Maldonado SG, Bethan B, Peter E, Schmitz O, Liebenberg V, Schatz P.分析前血液和血浆代谢变化的质量标记。临床医学杂志,2014;60(2):399-412。
Pinto J, Domingues MRM, Galhano E, Pita C, do canalu Almeida M, carira IM, Gil AM。人体血浆在处理和储存过程中的稳定性:对核磁共振代谢组学的影响。分析师2014;139(5):1168 - 77。
Sheiner LB, Beal SL.药代动力学的贝叶斯个体化:简单实现及与非贝叶斯方法的比较。中华药理学杂志;2002;31(3):344 - 344。
Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, Kern R, Picus M, Hoyer S, van Kerkwijk MH, Brett M, Haldane A, del Río JF, Wiebe M, Peterson P, g
熊猫开发团队,T.: panda -dev/ Pandas: Pandas。https://doi.org/10.5281/zenodo.3509134.
致谢
我们感谢Morgan Panitchpakdi和Shirley Tsunoda在解释等离子体数据集方面的帮助[41]。
资金
这项研究没有得到外部资助。维也纳大学开放获取基金。
作者信息
作者及单位
贡献
概念化由MG和JZ完成。资金由CG和JZ获得。方法由MG和JB开发。软件由MG开发。MG和CB进行形式分析。调查是由MG和JB完成的。资源由CG和JZ分配。验证由MG、JB和CB完成。本研究由CG和JZ指导。可视化由MG完成。 Original draft was written by MG and JZ. Review and editing of the manuscript was done by all authors. All authors read and approved the final manuscript.
相应的作者
道德声明
伦理批准并同意参与
不适用。
发表同意书
不适用。
相互竞争的利益
作者声明没有利益冲突。
额外的信息
出版商的注意
伟德体育在线施普林格·自然对已出版的地图和机构关系中的管辖权要求保持中立。
补充信息
附加文件1
.补充图、表和方程式。
权利和权限
开放获取本文遵循知识共享署名4.0国际许可协议,该协议允许以任何媒介或格式使用、共享、改编、分发和复制,只要您适当地注明原作者和来源,提供知识共享许可协议的链接,并注明是否进行了更改。本文中的图像或其他第三方材料包含在文章的知识共享许可协议中,除非在材料的署名中另有说明。如果材料未包含在文章的知识共享许可中,并且您的预期用途不被法律法规允许或超过允许的用途,您将需要直接获得版权所有者的许可。如欲查阅本许可证副本,请浏览http://creativecommons.org/licenses/by/4.0/.创作共用公共领域免责声明(http://creativecommons.org/publicdomain/zero/1.0/)适用于本文中提供的数据,除非在数据的信用额度中另有说明。
关于本文
引用本文
Gotsmy, M., Brunmair, J., b
收到了:
接受:
发表:
DOI:https://doi.org/10.1186/s12859-022-04918-1
关键字
- 代谢组学
- 手指出汗
- 血浆
- PKM
- PQN