- 研究gydF4y2Ba
- 开放获取gydF4y2Ba
- 发表:gydF4y2Ba
含羞草:共表达的混合模型,以检测调节相互作用的调节剂gydF4y2Ba
分子生物学算法gydF4y2Ba体积gydF4y2Ba5gydF4y2Ba、物品编号:gydF4y2Ba4gydF4y2Ba(gydF4y2Ba2010gydF4y2Ba)gydF4y2Ba
摘要gydF4y2Ba
背景gydF4y2Ba
功能相关基因倾向于在多种条件和/或组织类型中表达模式相关。因此,共表达网络常用于研究基因的功能群。特别是,当其中一个基因是转录因子(TF)时,基于共表达的相互作用被谨慎地解释为直接的调控相互作用。然而,任何特定的TF,更重要的是,任何特定的调节相互作用,可能只在实验条件的一个子集中活跃。此外,调节相互作用的表达样本子集可以通过存在或不存在修饰基因来标记,例如翻译后修饰TF的酶。当计算整体表达相关性时,这种微妙的调控相互作用被忽略了。gydF4y2Ba
结果gydF4y2Ba
在这里,我们提出了一种新的混合建模方法,其中一个tf基因对被认为是显著相关的(未知系数)在一个(未知的)表达样本子集。使用最大似然方法估计模型的参数。估计的表达样品的混合物,然后挖掘,以确定基因潜在调节tf -基因的相互作用。我们已经用合成数据验证了我们的方法,并在奶牛、酵母和人类的四个生物学案例中进行了验证。gydF4y2Ba
结论gydF4y2Ba
如前所述,虽然在某些方面存在局限性,但这项工作代表了一种挖掘表达数据和检测调节相互作用的潜在调节剂的新方法。gydF4y2Ba
背景gydF4y2Ba
真核生物的基因调控在很大程度上是在转录水平上进行的。许多功能相关的基因,例如,参与同一生物过程的途径成员,或其产物在物理上相互作用,往往具有相似的表达模式[gydF4y2Ba1gydF4y2Ba,gydF4y2Ba2gydF4y2Ba]。事实上,共表达已被广泛用于推断功能相关性[gydF4y2Ba3.gydF4y2Ba- - - - - -gydF4y2Ba6gydF4y2Ba]。已经提出了各种度量来量化相关表达,如Pearson和Spearman相关性[gydF4y2Ba2gydF4y2Ba]和相互信息[gydF4y2Ba5gydF4y2Ba]。然而,这些指标是对称的,它们既不提供因果关系,也不区分间接关系。例如,两个共表达的基因可能是共同调控的,或者一个可以直接或间接地调控另一个。gydF4y2Ba
转录调控的一个关键组成部分依赖于转录因子(TF)蛋白与靶基因相对附近的短DNA位点的序列特异性结合[gydF4y2Ba7gydF4y2Ba]。如果在共表达的两两分析中,其中一个基因是TF,那么通常假定因果关系是由TF直接指向另一个基因的。在没有这些资料的情况下,额外的后处理步骤[gydF4y2Ba5gydF4y2Ba]可以用来推断具有相关表达的基因对之间的方向性。此外,一阶条件独立度量[gydF4y2Ba4gydF4y2Ba已经被提出专门检测直接相互作用。gydF4y2Ba
虽然TF是转录的主要引擎,但它们的活性取决于其他几种蛋白质,如修饰酶和辅助因子,它们直接或间接地与TF相互作用以促进其活性。例如,TF CREB的活性取决于许多翻译后修饰,最值得注意的是蛋白激酶a对Ser133的磷酸化[gydF4y2Ba8gydF4y2Ba]。此外,对于许多TF, TF的活性可能仅限于特定的细胞类型和/或实验条件。因此,使用大量基因表达数据来估计共表达和功能相关性的常见做法有两个主要局限性:(1)它包括不相关的表达样本,这会给共表达信号增加噪音;(2)它忽略了额外修饰基因的贡献,因此无法检测到那些修饰基因是基因调控网络的关键组成部分。gydF4y2Ba
为了推断TF活性对组蛋白修饰酶的依赖性,Steinfeld等人分析了敲除特定组蛋白修饰酶的酵母样品中TF调控因子(TF的假定靶标)的表达[gydF4y2Ba9gydF4y2Ba]。在另一项研究中,Hudson等人分析了两组奶牛的表达数据,一组是肌肉较少的野生型,另一组是TF肌肉生长抑制素突变型[gydF4y2Ba10gydF4y2Ba]。他们发现,肌生长抑制素与差异表达基因MYL2的共同表达在突变型和野生型表达组之间存在显著差异。这种差异共表达使他们检测到肌生长抑制素是致病性TF,尽管突变型和野生型之间肌生长抑制素基因本身的表达没有差异。在上述两个例子中[gydF4y2Ba9gydF4y2Ba,gydF4y2Ba10gydF4y2Ba],这两组表情都有很好的特征,并且是先验的。事实上,Hu等人已经提出了一种非参数检验方法来检测两组表达样本中的差异相关基因对[gydF4y2Ba11gydF4y2Ba]。然而,在没有提供表达样本的适当划分的情况下,如何检测这种差异共表达基因对尚不清楚,也无法从实验描述中推导出来。这个问题是一个重要的实际挑战,为大型表达纲要涵盖了许多不同的实验条件。迅速增长的表达纲要[gydF4y2Ba12gydF4y2Ba],提供了一个独特的机会,不仅可以识别共表达和功能相关的基因,还可以预测基因调节因子的推定修饰因子。gydF4y2Ba
对于一对基因,我们在一组条件/样本中有表达数据,我们假设条件存在一些分区,使得两个基因在一个分区中相关,而在另一个分区中不相关。在这里,我们提出了一种新的方法,“Mimosa”,检测表达样本的隐藏划分为相关和不相关的子集。如果发现这样的分区,则表明存在修饰基因,如TF修饰酶,这些修饰基因应该在相关和不相关的样本分区之间差异表达。换句话说,跨样本的表达载体与样本分割载体相关的基因是假定的修饰因子。样本划分来源于共表达数据的混合模型。采用极大似然估计方法对混合模型的自由参数进行估计。一旦获得混合参数,我们就可以计算样本的加权划分为相关集和不相关集。在随后的步骤中,我们检测相关和不相关样本之间差异表达的假定修饰基因。通过合成数据,我们发现含羞草能够对表达样本进行分割,并具有较高的检测修饰基因的准确性。我们进一步介绍了四种生物应用,一种在牛样品中,两种在酵母中,一种在人类B细胞中。 This work represents a novel approach to mine expression data and detect potential modulators of regulatory interactions.
方法gydF4y2Ba
共表达的混合建模gydF4y2Ba
数字gydF4y2Ba1gydF4y2Ba说明了该方法。输入数据,即表达式概要,是一个矩阵gydF4y2Ba米gydF4y2Ba[gydF4y2Ba我gydF4y2Ba,gydF4y2BakgydF4y2Ba在那里,基因被索引gydF4y2Ba我gydF4y2Ba= 1,2,…,gydF4y2BaNgydF4y2BaggydF4y2Ba,是行和表达式样本,由gydF4y2BakgydF4y2Ba= 1,2,…,gydF4y2BaNgydF4y2Ba年代gydF4y2Ba,是矩阵的列。gydF4y2Ba米gydF4y2Ba[gydF4y2Ba我gydF4y2Ba,gydF4y2BakgydF4y2Ba]表示基因的表达gydF4y2Ba我gydF4y2Ba在表达样本中gydF4y2BakgydF4y2Ba。所有行都归一化,均值为0,方差为1。对于每一对基因gydF4y2Ba我gydF4y2Ba和gydF4y2BajgydF4y2Ba,有gydF4y2BaNgydF4y2Ba年代gydF4y2Ba表达式值对的数据点,(gydF4y2Ba米gydF4y2Ba[gydF4y2Ba我gydF4y2Ba,gydF4y2BakgydF4y2Ba],gydF4y2Ba米gydF4y2Ba[gydF4y2BajgydF4y2Ba,gydF4y2BakgydF4y2Ba])。为方便记法,我们将数据点表示为(gydF4y2BaxgydF4y2BakgydF4y2Ba,gydF4y2BaygydF4y2BakgydF4y2Ba)。观察到的基因对数据集,(gydF4y2BaxgydF4y2BakgydF4y2Ba,gydF4y2BaygydF4y2BakgydF4y2Ba),假设是两个不同分布的混合物:不相关样本组(组)gydF4y2BaugydF4y2Ba)和相关样本组(组)gydF4y2BacgydF4y2Ba),每一个都有自己的概率分布;称这些为分布函数gydF4y2BapgydF4y2BaugydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba),gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba)。通过定义gydF4y2BapgydF4y2BaugydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba) =gydF4y2BapgydF4y2BaugydF4y2Ba(gydF4y2BaxgydF4y2Ba)gydF4y2BapgydF4y2BaugydF4y2Ba(gydF4y2BaygydF4y2Ba),gydF4y2BapgydF4y2BaugydF4y2Ba(·)为正态分布。gydF4y2Ba

这幅图说明了含羞草背后的直觉gydF4y2Ba。考虑一个TF基因gydF4y2BaXgydF4y2Ba还有一个潜在的靶基因gydF4y2BaYgydF4y2Ba。的表达式值gydF4y2BaXgydF4y2Ba和gydF4y2BaYgydF4y2Ba对于所有的表达式样本显示为热图和散点图。我们假设gydF4y2BaXgydF4y2Ba和gydF4y2BaYgydF4y2Ba表达仅在样本的未知子集(用“+”表示)中相关,而在其余样本(用“-”表示)中不相关。Mimosa计算样本的最大似然分割。然后根据样本分割,第三个基因gydF4y2BaZgydF4y2Ba用微分表达式表示两个分区之间可能有潜在的修饰符。准确地说,我们为每个样本分配一个分区概率,而不是二元分区。gydF4y2Ba
观察到的数据被视为这两组混合分数的随机抽样gydF4y2BafgydF4y2Ba定义为属于不相关组的数据点的百分比。一个数据点(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba)是gydF4y2BapgydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba) =gydF4y2Baf pgydF4y2BaugydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba) + (1 -gydF4y2BafgydF4y2Ba)gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba)。在本分析中,我们假设不相关的分布为正态分布,因此,gydF4y2Ba

我们推导出相关数据的分布,gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba)通过假设存在一些(gydF4y2BaugydF4y2Ba,gydF4y2BavgydF4y2Ba)与(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba(通过旋转一个角度)坐标系gydF4y2BaθgydF4y2Ba,这样gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2BaugydF4y2Ba,gydF4y2BavgydF4y2Ba) =gydF4y2Ba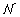 (gydF4y2BaugydF4y2Ba,gydF4y2BaσgydF4y2BaugydF4y2Ba)gydF4y2Ba(gydF4y2BavgydF4y2Ba,gydF4y2BaσgydF4y2BavgydF4y2Ba)。在这里,gydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaσgydF4y2Ba)是均值和方差均为零的高斯分布gydF4y2BaσgydF4y2Ba2gydF4y2Ba。(的坐标变换)gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba)坐标到(gydF4y2BaugydF4y2Ba,gydF4y2BavgydF4y2Ba)坐标为:gydF4y2BaugydF4y2Ba=gydF4y2BaxgydF4y2Ba因为gydF4y2BaθgydF4y2Ba-gydF4y2BaygydF4y2Ba罪gydF4y2BaθgydF4y2Ba和gydF4y2BavgydF4y2Ba=gydF4y2BaxgydF4y2Ba罪gydF4y2BaθgydF4y2Ba+gydF4y2BaygydF4y2Ba因为gydF4y2BaθgydF4y2Ba。变换的雅可比矩阵是1,所以我们有gydF4y2Ba
(gydF4y2BaugydF4y2Ba,gydF4y2BaσgydF4y2BaugydF4y2Ba)gydF4y2Ba(gydF4y2BavgydF4y2Ba,gydF4y2BaσgydF4y2BavgydF4y2Ba)。在这里,gydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaσgydF4y2Ba)是均值和方差均为零的高斯分布gydF4y2BaσgydF4y2Ba2gydF4y2Ba。(的坐标变换)gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba)坐标到(gydF4y2BaugydF4y2Ba,gydF4y2BavgydF4y2Ba)坐标为:gydF4y2BaugydF4y2Ba=gydF4y2BaxgydF4y2Ba因为gydF4y2BaθgydF4y2Ba-gydF4y2BaygydF4y2Ba罪gydF4y2BaθgydF4y2Ba和gydF4y2BavgydF4y2Ba=gydF4y2BaxgydF4y2Ba罪gydF4y2BaθgydF4y2Ba+gydF4y2BaygydF4y2Ba因为gydF4y2BaθgydF4y2Ba。变换的雅可比矩阵是1,所以我们有gydF4y2Ba

有三个未知数,{gydF4y2BaθgydF4y2Ba,gydF4y2Ba年代gydF4y2BaugydF4y2Ba,gydF4y2Ba年代gydF4y2BavgydF4y2Ba}。的形式有两个自然的限制gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba);即gydF4y2Ba


将这两个约束应用于eqn。(2),并假设gydF4y2BaσgydF4y2BaugydF4y2Ba≠gydF4y2BaσgydF4y2BavgydF4y2Ba,我们有gydF4y2Ba

式中-1≤gydF4y2BaαgydF4y2Ba≤1是混合模型的自由参数,控制相关分布的纵横比。不失一般性的让gydF4y2BaσgydF4y2BavgydF4y2Ba>gydF4y2BaσgydF4y2BaugydF4y2Ba;然后说到gydF4y2BaαgydF4y2Ba我们有gydF4y2BaσgydF4y2BaugydF4y2Ba2gydF4y2Ba= (1 - |gydF4y2BaαgydF4y2Ba|)和gydF4y2BaσgydF4y2BaugydF4y2Ba2gydF4y2Ba= (1 + |gydF4y2BaαgydF4y2Ba|)。请注意,gydF4y2BaαgydF4y2Ba< 0对应正相关数据(gydF4y2BaθgydF4y2Ba=gydF4y2BaπgydF4y2Ba/ 4)和gydF4y2BaαgydF4y2Ba> 0对应负相关数据(gydF4y2BaθgydF4y2Ba= -gydF4y2BaπgydF4y2Ba/ 4)。定义的宽高比gydF4y2BargydF4y2Ba≡gydF4y2BaσgydF4y2BavgydF4y2Ba/gydF4y2BaσgydF4y2BaugydF4y2Ba1、我们有gydF4y2BaαgydF4y2Ba| = (gydF4y2BargydF4y2Ba2gydF4y2Ba1) / (gydF4y2BargydF4y2Ba2gydF4y2Ba+ 1)。综上所述,混合模型有两个自由参数(gydF4y2BafgydF4y2Ba,gydF4y2BaαgydF4y2Ba),它决定了观测数据中不相关点的比例和相关数据分布的纵横比。gydF4y2Ba
观测数据的对数似然为gydF4y2Ba

我们最大化gydF4y2BalgydF4y2Ba在开源Gnu科学库中使用准牛顿-拉夫森函数优化例程进行数值计算gydF4y2Bahttp://www.gnu.org/software/gslgydF4y2Ba。得到的参数估计为gydF4y2Ba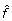 和gydF4y2Ba
和gydF4y2Ba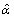 。gydF4y2Ba
。gydF4y2Ba
对于每个选择的基因对,我们计算每个样本属于相关组的概率。为gydF4y2BakgydF4y2BathgydF4y2Ba样本,它由gydF4y2Ba

这个概率向量等价于样本集的加权划分。根据修饰基因与载体的相关性选择修饰基因gydF4y2Ba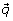 。我们使用基于预期总体数、平均值和方差的t检验来计算这种相关性(见下文)。当计算可行时,我们使用非参数相关度量,如肯德尔Tau。gydF4y2Ba
。我们使用基于预期总体数、平均值和方差的t检验来计算这种相关性(见下文)。当计算可行时,我们使用非参数相关度量,如肯德尔Tau。gydF4y2Ba
加权t统计量gydF4y2Ba
给定两个向量:(1)gydF4y2Ba向量表示每个样本的划分概率,(2)表达式向量gydF4y2Ba 原则上,对于潜在修饰基因的所有样本,我们可以仅根据划分概率将表达样本划分为两部分,然后使用t统计量或替代非参数检验比较两部分中的表达值。然而,这种方法需要任意选择分区概率阈值来对样本进行分区。相反,我们使用了加权版的t统计量,避免了任意阈值的需要。标准t统计量要求两个分区中的每一个都有三个参数:两个样本均值、两个样本标准差和两个样本大小。我们用加权和计算了所有这些参数。例如,相关分区的样本均值,gydF4y2BaμgydF4y2BacgydF4y2Ba,可估计为gydF4y2Ba
原则上,对于潜在修饰基因的所有样本,我们可以仅根据划分概率将表达样本划分为两部分,然后使用t统计量或替代非参数检验比较两部分中的表达值。然而,这种方法需要任意选择分区概率阈值来对样本进行分区。相反,我们使用了加权版的t统计量,避免了任意阈值的需要。标准t统计量要求两个分区中的每一个都有三个参数:两个样本均值、两个样本标准差和两个样本大小。我们用加权和计算了所有这些参数。例如,相关分区的样本均值,gydF4y2BaμgydF4y2BacgydF4y2Ba,可估计为gydF4y2Ba ,在那里gydF4y2Ba
,在那里gydF4y2Ba 为相关样本的加权数。类似地,相关分区的标准差,gydF4y2BaσgydF4y2BacgydF4y2Ba,由gydF4y2Ba
为相关样本的加权数。类似地,相关分区的标准差,gydF4y2BaσgydF4y2BacgydF4y2Ba,由gydF4y2Ba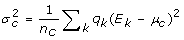 。gydF4y2Ba
。gydF4y2Ba
生成合成数据gydF4y2Ba
生成一个tf基因修饰子三联体gydF4y2BafgydF4y2Ba和gydF4y2BaαgydF4y2Ba我们执行了以下步骤。我们首先通过从正态分布中随机抽样来独立创建修饰符和TF表达式数据。对于给定的gydF4y2BafgydF4y2Ba,我们确定修饰符表达式的阈值gydF4y2Ba米gydF4y2Ba因此,低于此阈值的TF和基因被推定为不相关,高于此阈值的TF和基因被推定为相关。的价值gydF4y2Ba米gydF4y2Ba*估计为gydF4y2Ba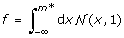 。我们生成的基因表达值如下。让gydF4y2Ba米gydF4y2Ba的修饰语表达gydF4y2BakgydF4y2BathgydF4y2Ba样本。如果gydF4y2BaM < MgydF4y2Ba*,然后该样本的基因表达值,gydF4y2BaygydF4y2BakgydF4y2Ba,是从正态分布(不相关分布)中得出的。如果gydF4y2Ba米gydF4y2Ba≥gydF4y2Ba米*gydF4y2Ba,则从均值-的高斯分布中得出基因的表达值gydF4y2BaαxgydF4y2BakgydF4y2Ba方差(1 -gydF4y2BaαgydF4y2Ba2gydF4y2Ba),gydF4y2BaxgydF4y2BakgydF4y2Ba的TF的表达式值是gydF4y2BakgydF4y2BathgydF4y2Ba样本。后一步源于相关数据的共表达式分布可以写成gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba) =gydF4y2BapgydF4y2BaugydF4y2Ba(gydF4y2BaxgydF4y2Ba)gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2Bay | xgydF4y2Ba),gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2Bay | xgydF4y2Ba)是均值为-的高斯分布gydF4y2BaαxgydF4y2Ba方差(1 -gydF4y2BaαgydF4y2Ba2gydF4y2Ba)。gydF4y2Ba
。我们生成的基因表达值如下。让gydF4y2Ba米gydF4y2Ba的修饰语表达gydF4y2BakgydF4y2BathgydF4y2Ba样本。如果gydF4y2BaM < MgydF4y2Ba*,然后该样本的基因表达值,gydF4y2BaygydF4y2BakgydF4y2Ba,是从正态分布(不相关分布)中得出的。如果gydF4y2Ba米gydF4y2Ba≥gydF4y2Ba米*gydF4y2Ba,则从均值-的高斯分布中得出基因的表达值gydF4y2BaαxgydF4y2BakgydF4y2Ba方差(1 -gydF4y2BaαgydF4y2Ba2gydF4y2Ba),gydF4y2BaxgydF4y2BakgydF4y2Ba的TF的表达式值是gydF4y2BakgydF4y2BathgydF4y2Ba样本。后一步源于相关数据的共表达式分布可以写成gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2BaxgydF4y2Ba,gydF4y2BaygydF4y2Ba) =gydF4y2BapgydF4y2BaugydF4y2Ba(gydF4y2BaxgydF4y2Ba)gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2Bay | xgydF4y2Ba),gydF4y2BapgydF4y2BacgydF4y2Ba(gydF4y2Bay | xgydF4y2Ba)是均值为-的高斯分布gydF4y2BaαxgydF4y2Ba方差(1 -gydF4y2BaαgydF4y2Ba2gydF4y2Ba)。gydF4y2Ba
结果与讨论gydF4y2Ba
合成数据gydF4y2Ba
给定一对混杂了相关和不相关样本的基因,以及一个表达与这两类样本相关的修饰基因,我们测试了我们的方法是否能检测出正确的修饰基因,这隐含地要求正确识别样本分区。仿真的细节在§Methods中提供。我们生成了1500个不重叠的TF-Gene-Modifier三胞胎,对于三胞胎中的每个基因,我们基于一个基础模型生成了300个样本的表达数据,参数化为gydF4y2BafgydF4y2Ba和gydF4y2BaαgydF4y2Ba。我们选择了一系列参数,并测试了这些参数对方法精度的影响。直觉上,含羞草最适合gydF4y2BafgydF4y2Ba的值在1/2附近gydF4y2BaαgydF4y2Ba接近±1。五种不同的值gydF4y2BafgydF4y2Ba所选择的大致包含的价值gydF4y2BafgydF4y2Ba= 0.5。作为gydF4y2BaαgydF4y2Ba不影响Mimosa对数据样本的划分能力,我们只选择正数gydF4y2BaαgydF4y2Ba。的三个值gydF4y2BaαgydF4y2Ba根据其相应的长宽比进行选择(见§Methods);即宽高比为2,3,5。不足为奇的是,当纵横比低于2时,含羞草的性能会恶化,也就是说,即使是相关样本(未显示)的相关性也非常差。每个参数箱包含100个TF-Gene-Modifier三胞胎(15箱× 100三胞胎/箱= 1500三胞胎,3 × 1500三胞胎= 4500个基因)。对于1500对TF-Gene中的每一对,我们应用含羞草(Mimosa)估计样本划分,然后根据其划分表达值的加权t检验p值对所有4500个基因进行排序(见§Methods)。对于每个二维垃圾箱(gydF4y2BafgydF4y2Ba和gydF4y2BaαgydF4y2Ba值),我们计算了箱子中100对TF-Gene对的正确修饰符的中位数排名(在4500个候选中)。我们还计算了100对tf基因对中正确修饰符排名最高的比例。gydF4y2Ba
如表所示gydF4y2Ba1gydF4y2Ba, Mimosa以高精度检测正确的样本分区和正确的修饰符。总的来说,在64.6%的情况下,正确的修饰语在最上面的位置被检测到。当90%的样品(上一列)中tf -基因对不相关时,则检测修饰子相对困难。即使这样,如果相关性很强(宽高比为5),那么含羞草仍然可以以非常高的精度检测到修饰语。注意,中位数排名最高的,为215gydF4y2BaαgydF4y2Ba= 0.6和gydF4y2BafgydF4y2Ba= 0.9 bin,当表示为4500个候选人中的百分位数时,仅为215/4500 = 4.8%。gydF4y2Ba
牛数据的应用gydF4y2Ba
Hudson等人比较了两种不同遗传杂交的表达谱gydF4y2BaPgydF4y2Ba和gydF4y2BaWgydF4y2Ba)。的gydF4y2BaPgydF4y2Ba型具有TF肌生长抑制素的突变形式,导致TGF-的失调gydF4y2BaβgydF4y2Ba途径和增加的肌肉量[gydF4y2Ba10gydF4y2Ba]。肌生长抑制素在这两种类型中的表达水平没有差异。他们进一步确定了两者之间差异表达的基因gydF4y2BaPgydF4y2Ba和gydF4y2BaWgydF4y2Ba,对于每一个这样的基因,以及920个假定的调节因子中的每一个,他们分别计算了基因和调节因子之间的表达相关性gydF4y2BaPgydF4y2Ba而在gydF4y2BaWgydF4y2Ba样本。基于两组样本中的这些成对相关性,他们确定了424对调控基因对,因此当使用来自的表达数据时,两者之间的表达相关性显著不同gydF4y2BaPgydF4y2Ba的表达式数据与表达式相关性进行了比较gydF4y2BaWgydF4y2Ba。这些数据为我们的方法提供了理想的测试平台。gydF4y2Ba
我们测试了含羞草将表达样本分成gydF4y2BaPgydF4y2Ba和gydF4y2BaWgydF4y2Ba没有任何先验知识。我们将424个调控基因对中的每一个进行混合建模,使用20个表达谱(10个为gydF4y2BaPgydF4y2Ba10分gydF4y2BaWgydF4y2Ba)载于[gydF4y2Ba10gydF4y2Ba]。这产生了424个分区概率向量gydF4y2Ba,每个长度为20(见§方法)。如果混合模型是有效的,我们期望{gydF4y2Ba问gydF4y2Ba1gydF4y2Ba、……gydF4y2Ba问gydF4y2Ba10gydF4y2Ba}(对应于gydF4y2BaPgydF4y2Ba)与{有显著差异gydF4y2Ba问gydF4y2Ba11gydF4y2Ba、……gydF4y2Ba问gydF4y2Ba20.gydF4y2Ba}(对应于gydF4y2BaWgydF4y2Ba),其中一个是高的,另一个是低的。我们使用Wilcoxon检验检验了这一假设,发现424对中有109对(26%)的p值≤0.05。因此,混合建模在许多情况下,即使表达式样本数量很少,也能正确地检索到隐藏的样本分区。gydF4y2Ba
酵母应用gydF4y2Ba
我们以前曾报道过一个数据库- ptm总机[gydF4y2Ba13gydF4y2Ba],其中包含510个酵母基因三胞胎,称为“MFG-triplets”,其中转录因子(F)调节基因(G),这种调节通过修饰酶(M)对F的翻译后修饰来调节。我们测试了含羞草是否能够正确划分一组表达样本并检测到修饰因子MgydF4y2Ba酿酒酵母gydF4y2Ba先前编译于[gydF4y2Ba14gydF4y2Ba来自18个不同的研究。这些实验包括细胞周期和各种应激条件。我们对每个F-G对应用含含水,然后计算样本划分概率向量的相关性(使用Kendall's Tau)gydF4y2Ba(参见§Methods)和所有6000个酵母基因的表达载体。然后我们计算了正确修饰语的排名(以百分位数为单位)。如图所示gydF4y2Ba2gydF4y2Ba结果发现,含羞草在前5%的样本中检测出了真正的修饰语(23%),比随机预期增加了5倍。gydF4y2Ba

在实验确定的510个TF-Gene-Modifier三胞胎中,从6000个候选修饰符中预测出的正确修饰符的百分位数分布gydF4y2Ba。含羞草在23%的情况下将正确的修饰语排在前5%。gydF4y2Ba
为了验证含羞草的大规模适用性,我们提取了全基因组ChIP-chip实验中检测到的所有酵母TF-Gene对[gydF4y2Ba15gydF4y2Ba]。为了减少要测试的基因对的数量,我们执行了以下过滤步骤。对于每一对,我们使用314个表达样本中的肯德尔Tau计算了它们的表达相关性。我们保留Kendall's Tau bonferroni校正的p值≤0.05的配对。在应用含羞草后,我们进一步过滤这个集合,只保留混合概率参数的情况gydF4y2BafgydF4y2Ba宽高比参数在0.45到0.55之间gydF4y2BaαgydF4y2Ba绝对值至少为0.8(高度相关)。对于得到的6960对TF-Gene,我们分别计算了相应的分割概率向量gydF4y2Ba。gydF4y2Ba
每个TF都有一组gydF4y2Ba问gydF4y2Ba-载体,每个载体对应TF的靶基因。生物学上,我们期望将样本划分为相关和不相关主要取决于TF是否活跃。如果是这样的话,那么在一组gydF4y2Ba问gydF4y2Ba-一个TF的向量。如图所示gydF4y2Ba3.gydF4y2Ba之间的肯德尔Tau相关gydF4y2Ba问gydF4y2Ba-具有相同TF的向量的分布确实明显倾向于正值,相对于随机选择的之间的相关性gydF4y2Ba问gydF4y2Ba向量。这个结果提供了一些证据gydF4y2Ba问gydF4y2Ba含羞草发现的矢量分区包含生物信息。gydF4y2Ba

之间的相关分布gydF4y2Ba问gydF4y2Ba-显示具有相同TF的向量,并将其与随机数向量的相关性分布进行比较gydF4y2Ba。所用数据取自酵母TF-Gene对;具体来说,是含羞草检测到的6960对酵母tf基因对。gydF4y2Ba
然后我们计算了每个基因表达载体之间的相关性gydF4y2Ba每一对gydF4y2Ba向量。每一对的“修饰因子”被认为是那些相关性符合bonferroni校正的加权t统计p值阈值0.05的基因。我们使用加权t统计量,而不是Kendall的Tau,主要是为了计算效率。然后,我们使用DAVID工具(DAVID .abcc.ncifcrf.gov)对由此获得的1356个假定修饰基因进行了功能富集分析。表格gydF4y2Ba2gydF4y2Ba图中显示了按注释具有该功能的输入基因的比例排序的富集(FDR < 5%)分子功能。最丰富的分子功能类别是“催化活性”,这与修饰酶的作用是一致的。即使我们为每个tf基因对选择单个最重要的修饰符,这种富集也会保持不变。需要做进一步的工作来分析检测到的特定修饰剂的生物学意义。gydF4y2Ba
申请STAT1gydF4y2Ba
转录因子STAT1在B细胞功能和B细胞癌中起关键作用[gydF4y2Ba16gydF4y2Ba]。STAT1活性是通过多种翻译后修饰来控制的[gydF4y2Ba17gydF4y2Ba- - - - - -gydF4y2Ba20.gydF4y2Ba]。我们试图用含羞草检测B细胞中STAT1的潜在上游调节剂。我们从[gydF4y2Ba21gydF4y2Ba]报告为STAT1靶标,并手动将其映射到50个转录本。我们还从[]获得了336个人类B细胞表达样本的摘要。gydF4y2Ba6gydF4y2Ba],其中包括基于HG-U95Av2 Affymetrix阵列的人类血液、癌症和细胞系样本。然后,我们将含羞草应用于所有由STAT1探针和对应于其中一个目标的探针组成的对。采用0.3≤的标准gydF4y2BafgydF4y2Ba≤0.7,|gydF4y2BaαgydF4y2Ba|≥0.8时,我们获得了10个与STAT1表达相关的靶标。检测到34个与分割载体表达相关的基因gydF4y2Ba(见方法),p值≈0。gydF4y2Ba
检测到的34种包括一些修饰酶,如激酶和磷酸酶,转录因子和辅助因子,以及膜受体。许多基因参与或与ifn - γ信号相关,ifn - γ信号是STAT1的主要激活因子[gydF4y2Ba22gydF4y2Ba],以及tgf - β和NF-kappaB信号,这两种信号在B细胞凋亡/存活中都很重要。检测到的几个基因,即GRK5和UBE21,已知在JAK-STAT信号传导中起作用。这些检测到的基因可能在影响STAT1活性的通路之间的串扰中发挥机制作用。然而,我们不能排除这些基因中的一些实际上在STAT1的下游或并行操作的可能性,在这种情况下,它们与分割载体相关gydF4y2Ba是由于一些共享和未检测到的上游调制器。我们总结了表中34个基因中的24个的这些发现gydF4y2Ba3.gydF4y2Ba。我们没有发现其他10个基因与STAT1有任何合理的联系。gydF4y2Ba
结论gydF4y2Ba
对于一对共表达基因(gydF4y2BaXgydF4y2Ba和gydF4y2BaYgydF4y2Ba),我们提出了一种混合建模方法来划分表达样本,以便检测样本的特定子集gydF4y2BaXgydF4y2Ba和gydF4y2BaYgydF4y2Ba表达式是强相关的。在某些情况下,这样的分割可能有助于检测其他基因之间可能调节的表达相关性gydF4y2BaXgydF4y2Ba和gydF4y2BaYgydF4y2Ba。这种电位调制器的特点是在两个样本分区中具有微分表达式。以前的几项调查与我们的工作密切相关。在[gydF4y2Ba10gydF4y2Ba]和in [gydF4y2Ba11gydF4y2Ba],给出两组表达样本,作者明确地寻找两组样本中表达相关性显著不同的基因对。另一种不同的方法,称为液体关联,明确地试图检测基因三胞胎(gydF4y2BaXgydF4y2Ba,gydF4y2BaYgydF4y2Ba,gydF4y2BaZgydF4y2Ba),其中变化之间的相关性gydF4y2BaXgydF4y2Ba和gydF4y2BaYgydF4y2Ba随值的变化而变化gydF4y2BaZgydF4y2Ba[gydF4y2Ba23gydF4y2Ba]。该方法基于调节基因表达隐式划分表达样本。相比之下,我们的方法在不了解调节基因的情况下对表达样本进行分区,并在随后的步骤中继续寻找调节基因。gydF4y2Ba
在全基因组应用中,例如在上面介绍的酵母应用中,原则上可以应用对数似然比(LLR)测试,其中混合模型具有自由的可能性gydF4y2BafgydF4y2Ba和gydF4y2BaαgydF4y2Ba参数与模型的似然进行比较,其中gydF4y2BafgydF4y2Ba= 0且仅gydF4y2BaαgydF4y2Ba是免费的。两个似然之比的对数可以用来评估基于a的划分的显著性gydF4y2BaχgydF4y2Ba2gydF4y2Ba分布。虽然使用LLR检验来评估混合模型的显著性很有吸引力,但我们发现我们的经验分布并不遵循agydF4y2BaχgydF4y2Ba2gydF4y2Ba分布(图gydF4y2Ba4gydF4y2Ba)。我们的下一个想法是通过随机排列表达式数据来使用经验推导的p值作为混合可能性。然而,可能性本身的经验分布在不同的基因对之间差异很大,因此我们不能使用全局分布。不幸的是,如果对每个基因对分别进行处理,则为充分解决p值所需的排列数量在计算上是不可行的。因此,作为一种实际的妥协,在全基因组酵母应用中,我们选择只考虑bonferroni校正的全局Kendall's Tau相关p值≤0.05的基因对。gydF4y2Ba

图中显示了(1)400个和1200个样本随机生成(正态,i.i.d)表达数据的对数似然比分布,排列了20000次;(2)gydF4y2BaχgydF4y2Ba2gydF4y2Ba1和2个自由度的分布gydF4y2Ba。“零”分布定义为gydF4y2BafgydF4y2Ba= 0,表示没有混合物。gydF4y2Ba
我们在该方法的第二阶段面临类似的挑战,其中,给定混合模型和样本划分概率向量gydF4y2Ba,我们根据它们的表达载体与的相关性来搜索调节基因gydF4y2Ba。对于大量的试验(候选调制器的数量),相关性的非参数检验,如肯德尔Tau,变得不可行的。因此,作为另一个实际的妥协,我们设计了加权t检验,它适用于合成数据。用于特定(gydF4y2BaXgydF4y2Ba,gydF4y2BaYgydF4y2Ba,gydF4y2BaZgydF4y2Ba)-三胞胎,我们使用肯德尔的Tau,但对于大规模应用,我们使用加权t统计量。需要进行更详细的研究,以仔细评估这些实际选择对该方法的准确性和有效性的影响。gydF4y2Ba
我们的混合模型可能在以下情况下最为有效:gydF4y2Ba10gydF4y2Ba],其中样本分区明显具有单个(未知)突变基因的特征。在大多数实际情况下,根据公开可用的表达式数据摘要,情况可能并非如此。真核生物中的调控关系有多个决定因素,即使该方法确实检测到“正确”的分区,也可能难以根据单个调节基因的差异表达来评估样本分区的生物学意义。gydF4y2Ba
总之,我们的工作为划分表达样本和检测一对基因之间表达相关性的潜在调节因子的问题提供了一种新的方法。虽然如上所述,这种方法在特定情况下可能是有效的,但统计和计算方面的挑战仍有待解决,需要做进一步的工作才能在大规模应用程序中利用这种方法。gydF4y2Ba
参考文献gydF4y2Ba
Tornow S, Mewes HW:蛋白质相互作用网络与基因表达相关的功能模块。核酸学报,2003,31(21):6283-9。10.1093 / nar / gkg838gydF4y2Ba
李建军,李建军,李建军,李建军。基于基因共表达网络的基因多态性研究。科学通报,2003,32(5):349 - 355。10.1126 / science.1087447gydF4y2Ba
冯德明,李建军,李建军,李建军,陈建军,陈建军,陈建军,陈建军,等。中国生物医学工程学报,2007,32(5):558 - 562。35数据库gydF4y2Ba
马文文,金杰:基于一阶条件独立的基因组共表达网络估计。中国生物医学工程学报,2004,5 (12):661 - 661 /gbgydF4y2Ba
马戈林,李建军,李建军,李建军,李建军,李建军,李建军,李建军,李建军。基于遗传算法的基因调控网络的构建。生物信息学,2006,7(增刊1):7-10.链接本文gydF4y2Ba
李建军,李建军,李建军,李建军,李建军,李建军,李建军,李建军。植物学报,2005,37(4):382-90。10.1038 / ng1532gydF4y2Ba
王晓明,王晓明,王晓明,等。人类转录因子的功能、表达和进化。农学通报,2009,10(4):252-63。10.1038 / nrg2538gydF4y2Ba
Khidekel N, hsih - wilson LC:“分子开关板”-蛋白质共价修饰及其对转录的影响。生物化学学报,2004,(2):1-7。10.1039 / b312466egydF4y2Ba
Steinfeld I, Shamir R, Kupiec M:酿酒酵母菌的全基因组分析表明染色质修饰因子对转录的影响。植物学报,2007,39(3):303-9。10.1038 / ng1965gydF4y2Ba
Hudson NJ, Reverter A, Dalrymple BP:表达数据的差异连线分析正确识别包含因果突变的基因。计算机工程学报,2009,35 (5):11000382 - 11371 /journal.pcbi.1000382gydF4y2Ba
胡仁,邱晓霞,郭志强,郭志强,等。基因筛选中基因间相关性变化的微阵列分析。生物信息学杂志,2009,10:20-10.1186/1471-2105-10-20gydF4y2Ba
Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R: NCBI GEO:高通量功能基因组数据档案。核酸学报,2009,D885-90。37个数据库gydF4y2Ba
李建军,李建军,李建军,等。转录因子翻译后修饰数据库的研究进展。核酸学报,2009,D66-71。37个数据库gydF4y2Ba
陈国强,陈建军,陈建军,等。基于cogrim模型的基因聚类研究。中国生物医学工程学报,2007,32 (1):559 - 559gydF4y2Ba
Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA:真核生物基因组转录调控密码。自然科学,2004,31(7):99-104。10.1038 / nature02800gydF4y2Ba
林国强,Mahajan S, Frank DA: STAT信号在白血病发病机制和治疗中的作用。中华肿瘤杂志,2000,19(21):2496-504。10.1038 / sj.onc.1203486gydF4y2Ba
温志,钟志,Darnell JJE: Stat1和Stat3转录的最大激活需要酪氨酸和丝氨酸的磷酸化。细胞生物学杂志,1995,32(2):241-50。10.1016 / 0092 - 8674 (95) 90311 - 9gydF4y2Ba
Rogers RS, Horvath CM, Matunis MJ: STAT1的SUMO修饰及其在pias介导的基因激活抑制中的作用。生物化学学报,2003,32(3):391 - 397。10.1074 / jbc.M301344200gydF4y2Ba
Kramer OH, Knauer SK, Zimmermann D, Stauber RH, Heinzel T:组蛋白去乙酰化酶抑制剂和羟基脲调控细胞周期并协同诱导细胞凋亡。中华肿瘤杂志,2008,27(6):732-40。10.1038 / sj.onc.1210677gydF4y2Ba
Soond SM, Townsend PA, Barry SP, Knight RA, Latchman DS, Stephanou A: ERK和F-box蛋白betaTRCP靶点STAT1的降解。生物化学学报,2008,29(3):387 - 398。10.1074 / jbc.M800384200gydF4y2Ba
Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffth OL, He A, Marra M, Snyder M, Jones S:基于染色质免疫沉淀和大规模平行测序的STAT1 DNA关联全基因组分析。地理学报,2007,4(8):651-7。10.1038 / nmeth1068gydF4y2Ba
Gough DJ, Levy DE, Johnstone RW, Clarke CJ: IFNgamma信号是否意味着JAK-STAT?细胞因子生长因子学报,2008,19(5-6):383-94。10.1016 / j.cytogfr.2008.08.004gydF4y2Ba
李克成:全基因组共表达动力学:理论与应用。美国国家科学促进会。中国生物医学工程学报,2002,29(6):387 - 398。10.1073 / pnas.252466999gydF4y2Ba
致谢gydF4y2Ba
SH由NIH R01-GM-085226资助,MH由NIH R21-GM-078203资助,LE由NIH T32-HG-000046资助,LS由NIH T32-HG-000046资助。这篇论文的一个版本发表在WABI 2009会议论文集上。gydF4y2Ba
作者信息gydF4y2Ba
作者及单位gydF4y2Ba
相应的作者gydF4y2Ba
额外的信息gydF4y2Ba
相互竞争的利益gydF4y2Ba
作者宣称他们没有竞争利益。gydF4y2Ba
作者的贡献gydF4y2Ba
SH, LE和MH构思了这个项目。MH开发并实现了该算法。LS帮助处理微阵列数据和一般统计问题。LE帮助进行STAT1分析。SH和MH撰写了手稿。gydF4y2Ba
马修·汉森,洛根·埃弗雷特对这项工作也做出了同样的贡献。gydF4y2Ba
作者提交的原始图片文件gydF4y2Ba
下面是作者提交的原始图片文件的链接。gydF4y2Ba
权利和权限gydF4y2Ba
开放获取gydF4y2Ba本文由BioMed Central Ltd.授权发表。这是一篇开放获取的文章,在知识共享署名许可(gydF4y2Bahttps://creativecommons.org/licenses/by/2.0gydF4y2Ba),允许在任何媒体上不受限制地使用、分发和复制,前提是正确引用原创作品。gydF4y2Ba
关于本文gydF4y2Ba
引用本文gydF4y2Ba
汉森,M,埃弗雷特,L,辛格,L。gydF4y2Baet al。gydF4y2Ba含羞草:共表达的混合模型,以检测调节相互作用的调节剂。gydF4y2BaMol BiolgydF4y2Ba5gydF4y2Ba, 4(2010)。https://doi.org/10.1186/1748-7188-5-4gydF4y2Ba
收到了gydF4y2Ba:gydF4y2Ba
接受gydF4y2Ba:gydF4y2Ba
发表gydF4y2Ba:gydF4y2Ba
DOIgydF4y2Ba:gydF4y2Bahttps://doi.org/10.1186/1748-7188-5-4gydF4y2Ba
关键字gydF4y2Ba
- 表达的相关性gydF4y2Ba
- 正确的修改器gydF4y2Ba
- 表达式示例gydF4y2Ba
- 分区向量gydF4y2Ba
- 混合建模方法gydF4y2Ba